RESUMEN
La polimiositis (PM) es una de
las miopatías inflamatorias,
son trastornos caracterizados
patológicamente
por la presencia de infiltrados inflamatorios en el músculo estriado. La
principal manifestación clínica de PM es la debilidad muscular proximal. Se desconoce la causa de la PM,
pero la evidencia actual sugiere que se trata de un trastorno autoinmune. La
debilidad muscular puede desarrollarse de forma repentina o más insidiosa durante un período de semanas a meses. El síntoma clásico de PM es la debilidad proximal, que puede manifestarse como dificultad para
sostener los brazos sobre la cabeza, subir escaleras o levantarse de una silla. La debilidad del
músculo estriado del esófago superior puede provocar disfagia, disfonía y
aspiración. Los
músculos de la pared torácica
pueden verse afectados, lo que lleva a compromisos
ventilatorios. La afectación del músculo
cardíaco puede
provocar arritmias e
insuficiencia cardíaca
congestiva.
La
dermatomiositis (DM) está estrechamente relacionada con la MP, y ambas se distinguen principalmente por la aparición de anomalías cutáneas características en la primera. PM y
DM pueden estar asociados con una variedad de tumores malignos. La MP también puede ocurrir como parte del espectro de otras enfermedades reumáticas como el lupus eritematoso
sistémico y la
enfermedad mixta del tejido conectivo. Además, la miopatía inflamatoria puede ser
causada por algunos medicamentos
(procainamida, D-penicilamina) y virus, sobre todo los retrovirus. Los
corticosteroides y los agentes inmunosupresores son los pilares de la terapia para PM. Los objetivos principales de la terapia son mejorar la fuerza y mejorar el funcionamiento físico. Muchos
pacientes requieren tratamiento
durante
varios
años.
Palabras
Claves: Polimiositis; Evaluación Diagnóstica;
Tratamiento; Medicamentos;
Fisioterapia.
ABSTRACT
Polymyositis (PM) is one of the inflammatory myopathies, they are disorders characterized
pathologically by the presence of inflammatory infiltrates in the striated muscle. The main clinical
manifestation of PM is proximal muscle
weakness. The cause of PM is unknown, but the current
evidence that it is an autoimmune disorder. Muscle weakness may develop suddenly or more insidiously over a period of weeks to months. The classic symptom of PM is proximal weakness, which can manifest itself as difficulty
in supporting the arms over the head, climbing stairs or
getting up from a chair. The weakness of the striated muscle
of the upper esophagus can cause dysphagia, dysphonia
and aspiration. The muscles of the chest wall can affect affected verses,
which leads to ventilatory compromises. Cardiac muscle involvement can cause arrhythmias and
congestive heart failure. Dermatomyositis (DM) is closely related to MP, and both are mainly
distinguished by the appearance of characteristic skin abnormalities in the first. PM and DM may
be associated with
a variety of malignant tumors. MP can also suffer as part of the spectrum of other
rheumatic diseases such as systemic lupus erythematosus and mixed connective tissue
disease. In addition, inflammatory myopathy can be caused by
some medications (procainamide,
D-penicillamine) and viruses, especially retroviruses. Corticosteroids and immunosuppressive
agents are the pillars of PM therapy. The main goals of therapy are to improve strength and
improve physical functioning. Many patients undergo treatment for
several years.
Key
Words:
Polymyositis; Diagnostic
Evaluation; Treatment; Medications; Physical Therapy.
Introducción.
La Dermatomiositis (DM) y la Polimiositis (PM) se clasifican como miopatías inflamatorias idiopáticas. Se
caracterizan clínicamente por
debilidad muscular proximal, elevaciones de las enzimas musculares séricas y adicionalmente en la DM por anormalidades de
la piel. Los mecanismos inmunes están involucrados en varios grados en la fisiopatogenia de PM
y DM. Ambos trastornos, tal como
se definen clínicamente, tienen tasas
de prevalencia
estimadas en aproximadamente 1 por 100,000
en la población general. Hay un predominio de mujer a hombre de aproximadamente 2: 1. La incidencia máxima en adultos es entre las edades de 40 y
50, pero cualquier grupo de edad puede verse afectado.
Los autores (Tymms &
Webb, 2015) expresan que la PM a menudo ocurre más
comúnmente en negros que
en
blancos. Los hallazgos histopatológicos típicos en el tejido muscular son infiltrados inflamatorios, compuestos principalmente por linfocitos T y macrófagos.
La DM se distingue de la
PM
por anormalidades de la piel, generalmente manifestadas por una erupción roja, escamosa y en forma de placa, sobre los nudillos, las muñecas, los codos, las rodillas
y los maléolos del tobillo. Además, a menudo hay lesiones violáceas en las áreas peri-orbitales y
del
tronco. (Strauss, Gonzalez-Buritica, Khamashta, &
Hughes, 2011). Típicamente habrá anormalidades
electromiográficas
(EMG), incluyendo
potenciales polifásicos, fibrilaciones espontáneas y descargas espontáneas de alta frecuencia. Los hallazgos característicos en la biopsia muscular son los de un proceso inflamatorio y necrotizante. Una clasificación propuesta para las miopatías inflamatorias idiopáticas es
la siguiente:
o PM idiopática primaria.
o DM idiopática primaria.
o PM o DM asociado con malignidad.
o Infancia PM
o DM.
La PM afecta los músculos esqueléticos
estriados, pero no los músculos lisos. Aunque se
desconoce el evento incitador de PM, se ha postulado que alguna lesión
micro vascular puede conducir a la liberación de autoantígenos musculares, que luego son presentados
a los linfocitos T por los macrófagos en el músculo. Los linfocitos
T que
se han activado luego proliferan y liberan citocinas como el interferón gamma y
la interleucina. El interferón gamma promueve una mayor
activación de macrófagos y la liberación de mediadores de la inflamación, como la IL-1 y el factor
de necrosis tumoral alfa. (Bohan & Peter, 2010)
La tasa de supervivencia a 5 años para
los pacientes tratados es del orden del 95%. Hasta
un tercio de los pacientes
con
PM pueden quedar con cierto
grado de debilidad muscular residual. Con el desarrollo de esta
investigación se busca dar
a conocer
los factores de riesgo de
esta
enfermedad, así como también, evaluar su diagnostico diferencial y tratamiento requerido en aras
de brindar apoyo para futuras
investigaciones acerca de este
tema.
Métodos y materiales.
Para el desarrollo de este proceso investigativo, se plantea como metodología la
encaminada hacia
una orientación científica
particular que se encuentra determinada por la necesidad de indagar en forma precisa y coherente una situación, en tal sentido (Davila, 2015)
define la metodología “como aquellos pasos previos que son seleccionados por el investigador
para lograr resultados favorables
que le ayuden a plantear
nuevas ideas”.(p.66)
Lo citado por el autor, lleva a entender que el desarrollo de la acción investigativa busca simplemente coordinar acciones
enmarcadas en una
revisión bibliográfica con el fin de complementar ideas previas relacionadas Polimiositis
factores de riesgo diagnostico diferencial y tratamiento, a través, de una revisión de
literatura, para así finalmente elaborar un cuerpo de consideraciones generales
que ayuden a ampliar el interés propuesto.
Tipo de Investigación
Dentro de
toda práctica investigativa, se precisan acciones
de carácter metodológico
mediante las cuales, se logra conocer
y proyectar los eventos posibles
que la determinan, así como
las características que
hacen del acto científico un proceso interactivo ajustado a
una realidad posible de ser interpretada. En este sentido, se
puede decir, que la presente
investigación corresponde
al
tipo documental, definido
por Castro (2016),
“se ocupa del estudio de
problemas planteados a nivel teórico, la información requerida para
abordarlos se encuentra básicamente en materiales impresos,
audiovisuales y /o electrónicos”.
(p.41).
En consideración a esta definición, la orientación metodológica permitió la oportunidad de cumplir con una serie de actividades inherentes a la revisión y
lectura de diversos documentos
donde se encontraron ideas explicitas relacionadas con los tópicos encargados de identificar a cada característica insertada
en
el estudio. Por lo tanto, se realizaron continuas interpretaciones con el claro propósito de revisar
aquellas apreciaciones o investigaciones propuestas por diferentes investigadores relacionadas con el tema de interés, para luego dar la respectiva argumentación a
los planteamientos, en función a las necesidades encontradas en
la indagación.
Fuentes Documentales
El análisis correspondiente
a las características que
predomina en el tema
seleccionado, llevan a incluir diferentes fuentes documentales encargadas de darle el respectivo apoyo y
en
ese sentido cumplir con la valoración de
los hechos a fin de generar nuevos criterios que
sirven de referencia a otros procesos investigativos. Para (CASTRO, 2016) las fuentes documentales incorporadas en la investigación documental o bibliográfica, “representa
la suma de materiales
sistemáticos que son revisados
en forma rigurosa y profunda para llegar a un análisis del
fenómeno”.(p.41). Por lo tanto, se procedió a cumplir con la realización de una lectura previa
determinada para encontrar
aquellos aspectos estrechamente
vinculados con el
tema,
con el fin de explicar mediante un desarrollo
las respectivas
apreciaciones generales
de importancia.
Técnicas
para la Recolección de la Información
La conducción de la investigación para
ser
realizada en función a las particularidades que
determinan a
los estudios documentales, tiene como fin el desarrollo de
un conjunto de acciones
encargadas de llevar a la selección de técnicas estrechamente vinculadas con las características del estudio. En tal sentido, (Bolívar, 2015), refiere, que es “una técnica particular para aportar ayuda a los
procedimientos de
selección de las
ideas
primarias y secundarias”. (p. 71).
Por
ello, se procedió a la utilización del subrayado, resúmenes, fichaje, como parte básica
para la revisión y selección de los documentos
que presentan el contenido teórico. Es decir, que
mediante la aplicación de
estas técnicas se
pudo llegar
a recoger
informaciones en cuanto a
la revisión bibliográfica de los diversos elementos encargados de orientar el proceso de investigación.
Tal
como lo expresa, (Bolívar, 2015)
“las técnicas documentales proporcionan
las herramientas
esenciales y determinantes para responder a los objetivos
formulados y llegar a resultados efectivos” (p. 58). Es decir, para responder con eficiencia a
las necesidades investigativas, se
introdujeron como técnica de recolección el método inductivo, que hizo posible
llevar a cabo una valoración de los hechos
de forma particular para llegar a la explicación desde una visión general.
Asimismo, se emplearon las técnicas de análisis de información para la realización de la
investigación que fue
ejecutada bajo la
dinámica de aplicar diversos elementos encargados de determinar el camino a
recorrer por el estudio, según, (Bolívar, 2015) las técnicas de procesamiento de datos en los estudios documentales “son las encargadas de
ofrecer al
investigador la visión o pasos que debe cumplir durante
su ejercicio, cada una de ellas debe estar en correspondencia con el nivel a emplear” (p. 123). Esto indica, que para
llevar a cabo el procesamiento de los datos obtenidos una vez aplicado las técnicas seleccionadas, tales como:
fichas de resumen, textual, registros descriptivos
entre otros, los mismos se deben ajustar al nivel
que ha sido seleccionado.
Resultados.
Ø Síntomas
Los
primeros síntomas suelen ser debilidad indolora de los músculos pélvicos y
de las extremidades inferiores proximales, lo que puede provocar dificultad para caminar y subir escaleras o levantarse
después de sentarse en una silla. Por lo general, los siguientes músculos
afectados son los del cuello y
la cintura escapular. El grado de debilidad puede variar de leve a casi parálisis cercana. La debilidad generalmente se desarrolla lentamente durante semanas o meses, aunque en
casos raros la
debilidad puede progresar más
rápidamente.
Otros síntomas de PM
incluyen:
ü Dificultad
para tragar
(disfasia).
ü Dificultad
para hablar.
ü Artralgia.
ü Fatiga.
ü Falta de aliento
El síntoma
predominante de PM es la
debilidad muscular. La
debilidad es simétrica y afecta los músculos
proximales de las extremidades y los flexores del cuello. La debilidad de los
músculos distales es rara, y
cuando está presente debe justificar la consideración de otro tipo de
miopatía, como la miositis por cuerpos de inclusión. Los pacientes con PM ocasionalmente pueden
experimentar dolor y
sensibilidad en los músculos, que pueden imitar los síntomas de la polimialgia reumática. (Strauss, Gonzalez-Buritica, Khamashta, & Hughes, 2011)
La afectación de los músculos estriados de la orofaringe y el esófago superior ocurre en el 10-15% de los pacientes, es un mal pronóstico indicador, y
puede conducir a disfagia, regurgitación y neumonía por aspiración. La enfermedad pulmonar intersticial ocurre en el 5-10% de
los pacientes. Además, puede haber disfunción ventilatoria debido a la afectación del diafragma
y los músculos intercostales. La afectación cardíaca suele ser asintomática, pero puede provocar
trastornos de la conducción, miocarditis o insuficiencia cardíaca congestiva.
El fenómeno
de Raynaud, la artritis no erosiva y los síntomas sistémicos
de rigidez matutina, fatiga, pérdida de peso y fiebre pueden estar
presentes durante el transcurso de
la PM. (Dalakas
& Hohlfed, 2003)
Ø Examen físico
La
debilidad simétrica de los músculos proximales es el hallazgo físico
más constante en
la PM. La sensibilidad muscular
puede estar presente
ocasionalmente, pero se preservan los
reflejos tendinosos profundos. El examen
sensorial suele ser
normal. El desgaste muscular o la atrofia pueden estar presentes en casos avanzados. El examen de los pulmones puede revelar crepitaciones inspiratorias secas. Se
puede observar disfonía con una calidad del habla nasal. La realización de
pruebas manuales
detalladas de la
fuerza muscular es de vital importancia. El
sistema de clasificación del Medical Research Council para la evaluación de la fuerza muscular es
ampliamente utilizado. La fuerza se clasifica según
la siguiente escala:
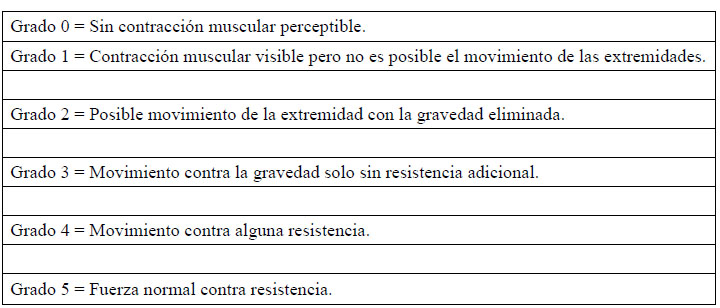
Otros hallazgos físicos pueden incluir sensibilidad y / o inflamación articular, erupción
cutánea y nódulos subcutáneos. (Olsen & Park,
2012)
Ø Criterios de diagnóstico y diagnóstico
diferencial.
Estudios de laboratorio
Enzimas musculares: los marcadores de laboratorio de lesiones musculares incluyen
elevaciones en los niveles sanguíneos de CK, aldolasa, AST, ALT
y LDH. Las elevaciones de CK ocurren en algún momento en la mayoría de los pacientes con PM, y CK es probablemente la
enzima más confiable para medir. Los niveles de CK a veces pueden ser normales al final de la enfermedad, o pueden permanecer
por encima de lo
normal incluso después de
que la fuerza muscular haya mejorado con el
tratamiento. (Greenberg,
2008)
Pruebas de imagen
Resonancia magnética (MRI)
A
medida que la resonancia magnética
se ha vuelto más sensible, se ha
vuelto más útil
en
el diagnóstico de
PM.
La resonancia magnética
de alta sensibilidad permite la detección de
inflamación muscular sutil temprano
en la enfermedad. La capacidad de obtener imágenes de
grandes áreas musculares en una extremidad puede ser útil para localizar
las áreas más anormales para una biopsia
muscular posterior. (Olsen & Park,
2012)
Electrodiagnóstico (EMG)
Al evaluar a un paciente
con PM, los estudios de EMG y de conducción nerviosa
pueden ayudar en el diagnóstico.
Los hallazgos de fibrilaciones
espontáneas en reposo o con
inserción de aguja, descargas espontáneas de alta frecuencia y ondas agudas positivas son las anormalidades
características. Las pruebas de EMG generalmente se realizan unilateralmente. La
biopsia muscular se puede
realizar
en
el lado contralateral para evitar el riesgo de
artefactos con aguja en
la muestra de biopsia. (Strauss,
Gonzalez-Buritica, Khamashta, & Hughes, 2011)
Biopsia muscular
La biopsia muscular es el procedimiento definitivo para establecer un diagnóstico de PM.
La
resonancia magnética y / o EMG pueden ayudar a identificar las áreas de mayor rendimiento
potencial para la biopsia. La biopsia muscular muestra fibras musculares en diversas etapas de
inflamación, necrosis y regeneración. Otros hallazgos patológicos incluyen infiltración endomisial
por células mononucleares, obliteración capilar, daño de células endoteliales y
aumento de la cantidad
de tejido conectivo. (Greenberg, 2008)
Ø Tratamiento
Los objetivos de la terapia son dobles: Para mejorar la debilidad muscular y para evitar el
desarrollo de enfermedades
extramusculares de los
órganos
vitales.
Se sabe que la presencia de enfermedad extramuscular de órganos vitales se
asocia con
peores resultados. En general, cuanto más grave es la enfermedad, menos
responde al tratamiento.
Los
pilares de la terapia son los corticosteroides y otros medicamentos
inmunosupresores. Las terapias complementarias no
farmacológicas también son
importantes. Estos incluyen regímenes
de ejercicio apropiados para la fuerza, medidas para prevenir la aspiración y
atención de apoyo
general.
Corticosteroides
Los corticosteroides como la prednisona son la primera línea
de terapia para PM. La dosis
inicial habitual es de 1 mg / kg / día de prednisona o su equivalente. Esta dosis generalmente se mantiene durante las primeras 6-8 semanas. La respuesta a la terapia
debe evaluarse cada 2 a 4
semanas controlando la fuerza muscular proximal, los niveles de enzimas musculares y
la funcionalidad del paciente. La fuerza muscular y las medidas funcionales son mejores indicadores de mejora que los niveles de
enzimas musculares.
Después de las primeras 6-8 semanas, debe comenzar una disminución lenta de los esteroides. El objetivo es disminuir gradualmente los esteroides o la dosis efectiva más baja posible durante un período de 9-12 meses. Una proporción significativa de pacientes no puede tratarse con esteroides solos, ya sea debido a los efectos secundarios, el control deficiente de la enfermedad o ambos. Muchos reumatólogos comienzan un agente inmunosupresor ahorrador de esteroides en el
momento
en que se inician los
esteroides, mientras que otros
prefieren reservar estos agentes para
pacientes que claramente han fallado en la monoterapia con corticosteroides. (Majithia & Harisdandkul, 2005)
Drogas ahorradoras
de esteroides
En un paciente que responde a los esteroides,
el objetivo es alcanzar la dosis más baja de esteroides que controle adecuadamente la enfermedad. Para
lograr este objetivo,
los agentes ahorradores de esteroides son necesarios en la
mayoría
de los pacientes con MP. El proceso de selección de
medicamentos ahorradores de
esteroides es empírico, aunque los más utilizados son
la azatioprina
(AZA) y el metotrexato (MTX).
AZA generalmente se administra por vía oral a una dosis de 1.5-3 mg / kg / día. El
medicamento puede no ser completamente efectivo por hasta 4 meses. Las náuseas, la
supresión de la médula ósea y
la hepatotoxicidad son los principales efectos adversos. Se requiere un monitoreo de laboratorio
regular de los recuentos sanguíneos completos y estudios de función hepática. (Dalakas & Hohlfed, 2003)
MTX puede
administrarse por vía
oral, subcutánea
o intramuscular.
MTX se administra una
vez a la semana en dosis que oscilan entre 10 y 40 mg. Las dosis superiores a 20 mg semanales se toleran mejor cuando se
administran por vía
parenteral. El efecto máximo de MTX se
observa
en
2 meses. La supresión de la médula ósea y
la
hepatotoxicidad son los principales efectos
adversos, y la dosis debe ajustarse a la baja ante la insuficiencia renal. Se requiere un monitoreo de laboratorio regular de los recuentos sanguíneos completos, pruebas de función hepática y creatinina sérica. (Joffe, 2013)
Algunos pacientes, después de una respuesta inicial a las terapias convencionales, pueden desarrollar actividad recurrente de la enfermedad. Otros
pacientes pueden no responder de manera óptima a las terapias convencionales. Aunque los
datos de eficacia son limitados, existen varias opciones
de tratamiento para pacientes en
las categorías
mencionadas.
Micofenolato de mofetilo.
El micofenolato de mofetilo se administra por vía oral en dosis de hasta 3 g
/ día. El medicamento puede no
ser máximo efectivo durante 3 meses. Generalmente es bien tolerado.
Los síntomas gastrointestinales y la leucopenia son los efectos adversos más comunes. (Majithia & Harisdandkul, 2005)
Inhibidores de la
calcineurina.
La ciclosporina A y el tacrolimus han demostrado cierta eficacia en el tratamiento de la
PM
refractaria. La ciclosporina A se administra por vía oral en dosis de hasta 150 mg dos veces al
día. La hipertensión y la nefrotoxicidad son los principales efectos adversos. La dosis óptima de tacrolimus no está clara. Un estudio utilizó dosis de 0.075 mg / kg / día en 2 dosis divididas, con normalización
de la fuerza en 5 de 8 pacientes. (Greenberg, 2008)
Inmunoglobulina intravenosa
En pacientes que son resistentes a los corticosteroides, especialmente donde hay una
progresión rápidamente progresiva o potencialmente
mortal, la inmunoglobulina intravenosa (IGIV) puede ser útil. La dosis inicial recomendada es de 2 g / kg. La mejora en la fuerza puede ser
evidente a los pocos días de
la primera infusión. Las infusiones repetidas a intervalos de 5-8 semanas pueden
ser necesarias para mantener
la respuesta. (Dalakas & Hohlfed, 2003)
Rituximab
El rituximab (RTX) es un anticuerpo monoclonal contra
las células B CD 20 positivas,
lo que provoca el agotamiento de estas células durante 6 meses o más. La dosis óptima de RTX en PM es desconocida. Un protocolo es usar 375 mg / m2, infundidos por vía
intravenosa una vez a la semana durante 4 semanas. Otro régimen de dosificación es administrar 2 infusiones quincenales
de 1
g (dosis total de 2 g) por vía intravenosa. Un ensayo multicéntrico, controlado con placebo de RTX en
pacientes con PM está
actualmente en
curso. RTX es generalmente bien tolerado. Las
reacciones a la perfusión generalmente
se pueden controlar con corticosteroides o
antihistamínicos. También hay un aumento
en el riesgo de infecciones en pacientes tratados con
RTX. (Levine, 2005)
Ciclofosfamida
La ciclofosfamida (CTX) es un agente
alquilante que es tóxico para las células linfopoyéticas. Tanto las células T como las células B productoras de anticuerpos se ven afectadas.
CTX tiene una considerable toxicidad hematológica y vesical, y es un potente inmunosupresor.
Por
lo tanto, CTX debe reservarse para
aquellos pacientes con PM que tuvieron múltiples fallas
con
otros agentes de segunda línea. Este medicamento puede ser más útil en pacientes con PM con enfermedad
pulmonar intersticial.
El medicamento puede administrarse por vía
intravenosa a 0.8-
1 g / m2 / mes durante varios meses. CTX también se puede administrar por vía oral a dosis de
1.5-2
mg / kg / día.
(Vencovský,
Machácek,
Studýnková, Kafková, & Bartůnková,
2009)
Conclusiones.
Durante el desarrollo del trabajo investigativo se
observó que la PM es un trastorno inflamatorio idiopático del músculo estriado que ocurre
más comúnmente en mujeres entre las edades de 50 y 70 años. La manifestación clínica predominante es la debilidad muscular proximal.
También se observó que puede haber afectación muscular adicional, como artritis
inflamatoria, fenómeno de Raynaud, miocarditis y
enfermedad pulmonar intersticial. Su diagnostico es detectado por
medio de las enzimas musculares séricas que
en
estos casos generalmente están elevadas
durante los períodos en que enfermedad está activa.
Las anomalías
características frecuentemente se ven en EMG y
MRI
muscular. Sin
embargo, el diagnóstico
definitivo se establece mediante
biopsia muscular. Una vez diagnosticada
la patología, los corticosteroides son la
base de la terapia, pero adicionalmente se
utilizan otros
agentes inmunomoduladores en el tratamiento
de esta enfermedad. La mayoría
de los pacientes responden a la terapia, aunque no
es infrecuente algún grado de
daño muscular a largo
plazo.
Bibliografía.
Bohan, A., & Peter, J. (2010). Polimiositis y dermatomiositis (segunda de dos partes) . Engl J
Med.
Bolívar, J.
(2015). Investigación
Documental. México. Pax. Castro,
J. (2016). Técnicas
Documentales. México. Limusa.
Dalakas, M.,
& Hohlfed, R. (2003). Polymyositis
and dermatomyositis.
. Lancet, 71–82. Davila, A. (2015). Diccionario de
Términos Científicos. . Caracas: Editorial Oasis.
Greenberg, S. (2008). Infl ammatory myopathies: Evaluation and management. Bogota: Semin
Neurologico.
Joffe, M. (2013). Drug therapy of the idiopathic inflammatory myopathies: predictors of response
to prednisone, asathioprine,
and methotrexate and a comparison of their efficacy. 79-87.
Levine, T. (2005). Rituximab en el tratamiento de la dermatomiositis: un estudio piloto de etiqueta abierta. Artritis Rheum.
.
Majithia, V., & Harisdandkul, V. (2005). Micofenolato mofetilo (CellCept): una terapia alternativa
para la miopatía inflamatoria autoinmune.
. Reumatología
(Oxford),
86–9.
Olsen, N., & Park, J. (2012). Miopatías inflamatorias: problemas en el diagnóstico
y manejo .
Arthritis Care Res. , 100-200.
Strauss,
K.,
Gonzalez-Buritica,
H.,
Khamashta,
M.,
&
Hughes, G. (2011).
Polymyositis- dermatomyositis:
a clinical review. . Postgrad Med J.
Tymms, K., & Webb, J. (2015). Dermatopolymyositis and other
connective tissue diseases: A
review of 105 cases. . J Rheumatol.
Vencovský, J.
J., Machácek, S.,
Studýnková, J.,
Kafková,
J., & Bartůnková, J.
(2009).
Cyclosporine A versus methotrexate in the treatment of polymyositis and dermatomyositis.
Scand J
Rheumatol. , 95–102.
