
Efrén Bryan
Barco Ramírez a; Verónica Ayling
Lama
Asínc b; Jorge Andrés Carbo
Palacio c; Freddy Leónidas Monge Paladines
d
Síndrome de McCune-Albright:
multiples
fracturas
patologicas en paciente con menarquia precoz
McCune-Albright syndrome: multiple pathological
fractures in patients with early
menarche
Revista Científica
Mundo de la Investigación
y el Conocimiento.
Vol. 3 núm. 4., diciembre,
ISSN: 2588-073X, 2019, pp. 586-605
DOI: 10.26820/recimundo/3.(4).diciembre.2019.586-605
URL: http://recimundo.com/index.php/es/article/view/683
Código UNESCO: 3205 Medicina Interna
Tipo de Investigación: Artículo
de Revisión
© RECIMUNDO; Editorial Saberes del
Conocimiento,
2019
Recibido: 15/09/2019 Aceptado: 23/11/2019 Publicado:
30/12/2019
Correspondencia: marthica12@msn.com
a. Médico; Investigador Independiente; Guayaquil, Ecuador; Efrenbarcoramirez@outlook.es b. Médico; Investigador Independiente; Guayaquil, Ecuador; aylinglamaasinc@gmail.com
c. Médico; Investigador Independiente; Guayaquil, Ecuador; jcarbop91@gmail.com
d. Médico; Investigador Independiente; Guayaquil, Ecuador; fmongepaladines_91@yahoo.com
RESUMEN
El síndrome de McCune-Albright, es una
entidad genética no hereditaria y poco frecuente que corresponde a trastorno
postzigótico producido por una mutación somática en el gen GNAS 1. La incidencia mundial es desconocida, pero se estima una frecuencia de un caso por cien mil a un caso por
millón de habitantes, esta entidad suele
componerse clásicamente
por displasia fibrótica poliostótica, manchas café
con
leche y alteraciones endocrinas; de esta ultima la
principal es la pubertad precoz que puede debutar con una menarquia precoz, sin embargo, se pueden asociar otras endocrinopatías. El diagnostico puede estar basado en la clínica
cuando se tienen dos de los tres componentes clásicos del síndrome, sin embargo, se pueden requerir de estudios endocrinos completos, pruebas de
imágenes, histopatología
y estudios genéticos cuando no se manifiesta de manera
clásica. El tratamiento para
el
síndrome de McCune-Albright
no es especifico, puede incluir
fármacos que controlen las
manifestaciones endocrinas, intervenciones
quirúrgicas para las endocrinopatías o fracturas derivadas
de la displasia fibrosa poliostótica.
Palabras Claves: Displasia Fibrótica Poliostótica; Manchas Café
con Leche;
Síndrome de
Mccune-Albright;
Menarquia Precoz.
ABSTRACT
The McCune-Albright syndrome
is a non-hereditary
and
rare genetic entity that corresponds
to a postzigotic disorder produced by a somatic mutation in the GNAS 1 gene. The global incidence
is unknown, but it’s estimated a frequency of one
case
per hundred thousand to one case per million inhabitants, this
entity is usually composed by polyostotic fibrotic dysplasia, café-au-lait
spots and endocrine alterations; of the latter the main
one is precocious puberty that can debut with precocious
menarche, however other endocrinopathies can be associated.
The diagnosis can be based on clinic when two of the three classic components of the
syndrome are present, however, complete endocrine studies, imaging tests and genetic studies may be required when
the disease doesn’t have a classical presentation. Treatment for McCune-Albright
syndrome is not specific, this may
include drugs that control
endocrine manifestations, surgical interventions
for endocrinopathies
or fractures
derived from polyostotic fibrous dysplasia.
Keywords: Polyostotic Fibrotic
Dysplasia; Café-Au-Lait Spots; Mccune-Albright Syndrome; Early
Menarche.
Introducción.
El síndrome de McCune-Albright constituye una
entidad poco común cuya exacta incidencia mundial es desconocida y en nuestro país la incidencia estimada y exacta es también
desconocida. La clínica puede incluir los componentes clásicos que
caracterizan a
este síndrome como son la displasia fibrosa poliostótica, pigmentación de
la piel bajo la forma de manchas café
con
leche y pubertad precoz; en este caso el diagnóstico puede
ser sospechado. La
expresión clínica depende de la
cantidad de células mutadas y órganos afectados, debido a
esto no siempre está presente
la tríada clásica y el diagnóstico puede ser difícil, pudiendo
así, ser infradiagnosticado.
Presentación de caso
Paciente femenino de 28 años, etnia mestiza de la provincia del Guayas que acudió en
enero
del 2016 al IESS sur (Valdivia)
APP:
menarquia a los 2 años de
edad( se trato con danazol 400mg/dia) prolapso mitra
fractura patológica de fémur a
la edad de 17 años la cual
fue tratada mediante
curetaje.
APF: no refiere
patologias de importancia paciente acude en enero del 2016 al IESS sur
valdivia por presentar claudicación de
la marcha y dolor a nivel de aductores derechos de
1 mes de
evolución el mismo
que exacerbaba al realizar maniobra de aducción
contra resistencia.
Exploración física
• Signos vitales: TA 140/80 mmhg FC: 78 lpm
FR:
21 T 36.5ª.C
• Cara y craneo: asimetría facial, dolor que exacerbaba a la palpación de la articulación temporomaxilar y durante la
masticación.
• Oftalmologica: hiporreflexia pupilar leve del ojo izquierdo y defecto pupilar aferente del mismo
ojo
• Cuello:
simetrico
• Torax: simetrico
• Abdomen: blando con
dolor en la palpacion
profunda
• El
resto de la exploracion fisica sin alteraciones
Laboratorio
• Leucocitos
(wbc): 7.75ul
• Hemoglobina
(hgb): 12.6 g/dl
• Hematocrito
(hct):
38.7ml/dl
• Volumen
corp. medio (mcv): 86.6 fl
• Conc.media
hemog. (mch): 28.2g/dl
• Conc.corp.media (mchc) de hemo: 32.6 g/dl
• Glucosa:
91mg/dl
• Urea: 23mg/dl
• Creatinina: 0.62 mg/dl
• Trigliceridos: 398mg/dl
• Colesterol: 293mg/dl
• Calcio total: 8.80
mg/dl
• IGf-1.: 111.00(U/ml)
• Lh
hormona luteinizante.:
1.96mUI/ml
• 17-beta
estradiol.:
904.60
pg/ml
• Parathormona:
20.90 pg/ml
Estudios imagenológicos
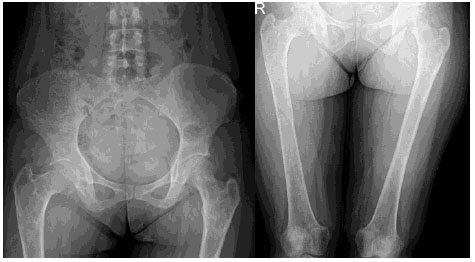
• Pelvis ósea:
Imágenes oseas endomedulares predominantemente
líticas que
comprometen también ambos fémures particularmente
cabezas y epífisis proximales. las lesiones expanden la cortical
ósea adelgazándola sin
destrucción de la misma.
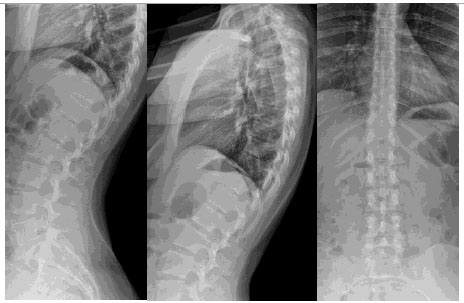
• Columna dorso
lumbar:
Cifosis dorsal levemente pronunciada, discreta rotoescoliosis dorso
lumbar.
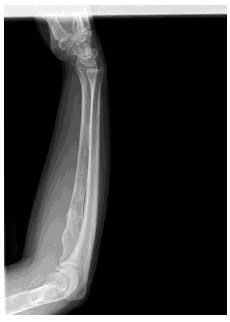
• Humero cubito y radio:
Se observan múltiples imágenes oseas endomedulares predominantemente líticas que
comprometen tercio distal de humero, tercio proximal y medial del radio, además de observarse fractura consolidada en tercio proximal del mismo, las lesiones expanden
la cortical ósea adelgazándola sin destrucción de la misma
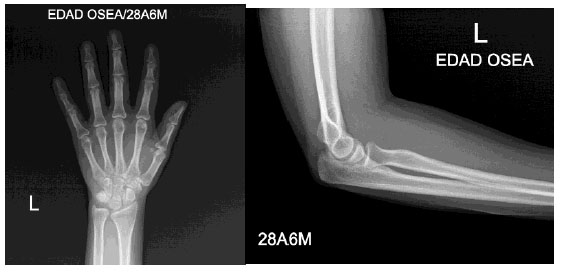
• Radiografía de edad
ósea
Radiografía de edad ósea no
dominante:
Según
el método de Greulich-Pyle en base a la maduración de huesos y muñeca, la edad
ósea corresponde a mayor
de 18 años.
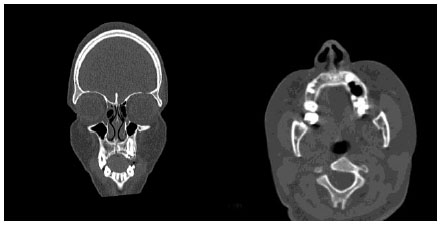
Tomografía cabeza
• Articulación
temporomandibular:
Presenta canino superior izquierdo
retenido por odontoma.
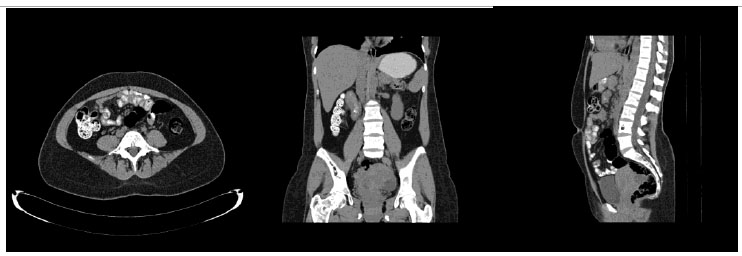
• Tomografía abdominal
En
ventana ósea se aprecian imágenes líticas en L3, L4 y L5; y a nivel de crestas iliacas y
femorales.
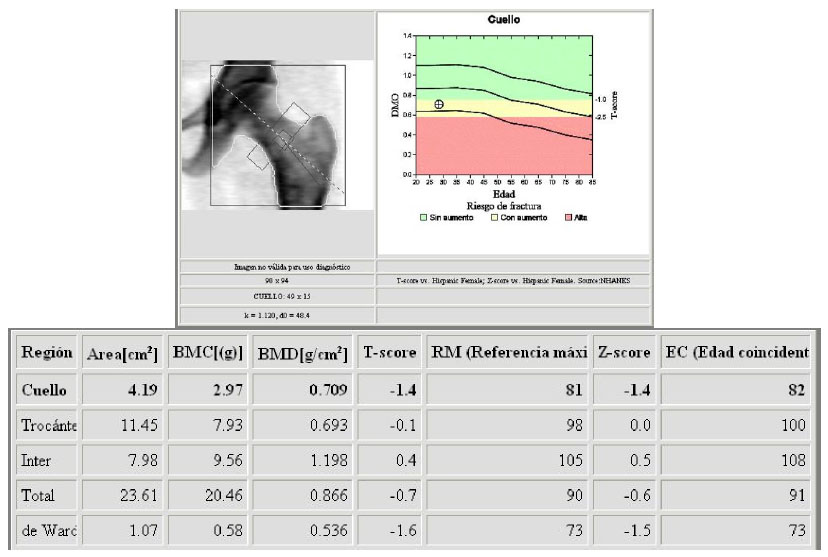
Densitometria
osea
Riesgo de fractura: aumentado.
Diagnóstico: osteopenia
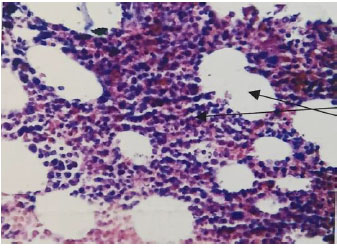
Biopsia puncion
Tejido de muestra: medula osea de cresta
iliaca.
Diagnóstico:
medula
ósea normocelular con maduracion
normoblastica.
Paciente es diagnosticada con
el
síndrome de MCcune Albright.
Discusión.
En el pasado, la descripción completa del síndrome de McCune Albright (SMA) se realizó de forma separada por los colaboradores de Donovan McCune y Fuller Albright, a pesar
de que previamente se reportaron casos parecidos. En el año 1936, D.
McCune relató el caso de
una niña con pigmentación cutánea con manchas café
con
leche, displasia fibrosa poliostótica, pubertad precoz e hipertiroidismo. Al año siguiente F. Albright
reunió 5 casos femeninos
similares, por lo cual en 1937 se logró obtener información más completa de esta entidad que tomo
el nombre de sus descubridores. (1) (2)
(3)
El síndrome de McCune-Albright
constituye una entidad poco común que asocia la tríada compuesta por displasia fibrosa poliostótica, pigmentación de la piel bajo la forma de manchas café con leche y pubertad precoz, sin embargo, se ha reconocido que otras endocrinopatías, incluidos el hipertiroidismo, el exceso de hormona de crecimiento (GH), la pérdida renal de fosfato con o sin raquitismo / osteomalacia y el síndrome de Cushing podrían encontrarse en asociación con la tríada original. La forma no clásica del SMA consta de solo dos de los tres componentes de la triada clásica. En raras ocasiones, otros órganos pueden estar involucrados (hígado, corazón, paratiroides, páncreas). La incidencia mundial del SMA es desconocida, pero se estima una frecuencia de 1 caso por cien mil a 1 caso por millón.
En el año 2012 en un estudio realizado en dos hospitales del Centro Medico Nacional Siglo XXI en Mexico D.F se valoro a 12 pacientes de entre 19 meses a 29 años de edad que presentaron displasia fibrotica poliostotica, manchas café con leche y alteraciones endocrinas,
siendo así una enfermedad poco común que se presenta con poca
frecuencia,
debutando en la infancia y afectando
a ambos sexos, sin embargo, por razones desconocidas posee
un claro predominio por
la población femenina (relación mujer/
varón de 10 a 1), no posee una
predilección por una raza o etnia específica. (2)
(4) (5) (6) (7) (8)
La
expresión clínica
depende de la cantidad de
células mutadas y órganos afectados. Por
lo tanto, la presentación puede
ser heterogéneo, que
implica varios órganos endocrinos y no endocrinos.
Puede ser de inicio temprano o tardío con evolución
lenta o rápida. Esta anormalidad
conduce a la
proliferación de
células osteoprogenitoras
indiferenciadas,
por lo tanto, hay aumento de la matriz fibrosa con tejido oseo. La
presentación clínica de
SMA
es altamente
variable, dependiendo de cuál de
los componentes del síndrome
predomina. La
forma clásica de SMA es más común en las mujeres y está definida por la tríada compuesta por displasia fibrosa poliostótica, endocrinopatía e hiperpigmentación de la piel, pero para instaurar el diagnóstico es aceptable
la asociación de
2 de los 3 hallazgos que componen la tríada clásica, ya
que en la mayoría de casos es un síndrome evolutivo. Por lo general, los signos y síntomas de pubertad
precoz
o displasia fibrosa
ósea debutan como presentación inicial, mientras que la pigmentación cutánea puede presentarse a nacer o poco después. La
pubertad precoz puede manifestarse como sangrado vaginal en las niñas o agrandamiento testicular en los niños. Otros síndromes endocrinos pueden estar
presentes, incluyendo hipertiroidismo, acromegalia
y síndrome de Cushing. El síndrome de Mazabraud, que también puede existir en asociación con SMA e
implica la aparición de mixomas
y, por lo general, displasia fibrosa poliostótica (9) (7)
(10)
El trastorno es el resultado de un trastorno postzigótico
que consiste en una mutación somática en el gen GNAS 1 en el locus 20q 13.1-13.2 , que
codifica la subunidad alfa de la
proteína G estimulante (Gsa). Las proteínas G acoplan los receptores de
superficie celular a
proteínas intracelulares para activar o inactivar cascadas de señalización. La proteína G
estimuladora normalmente se activa cuando una hormona u otro ligando se une al receptor de
superficie de la célula. La subunidad Gsa activada posteriormente se disocia del receptor, se une
a la adenililciclasa y estimula
un aumento en los niveles de adenosin monofostato cíclico intracelular
(cAMP) y posteriomente
existe falla en la
diferenciación del osteoblasto, y que
vincula a un incremento
en la resorción ósea por
parte los osteoclasto, este último proceso es
inducido por IL-6; a la vez el incremento celular del AMPc a nivel de los melanocitos en los
cuales está presente la mutación estimula la actividad de la tirosinasa, lo que hace que exista
una sobreproducción de melanina.
La subunidad Gsa se inactiva, se vuelve a asociar con el receptor y vuelve a ser disponible para reactivarse por medio de hormonas. Dado que el SMA resulta de
una mutación somática postzigótica y existe
un mosaicismo genético, todas las células hijas de la célula embrionaria en la que se produjo la mutación inicial también contienen la
mutación. Cuanto antes se produce la mutación en la embriogénesis, más extendida está la afectación del tejido. Las mutaciones tardías en la embriogénesis están más enfocadas y explican los casos leves en los que solo están presentes 2
o 3 de las características fenotípicas clásicas del síndrome. Si la mutación ocurre muy tarde en el desarrollo del tejido después de la diferenciación en una línea celular específica, entonces puede producirse un solo adenoma. Se han
informado mutaciones Gs alfa-activadoras en nódulos
tiroideos hiperfuncionantes aislados y en adenomas
somatotropos (2) (11) (4)
(9)
La
displasia fibrosa generalmente se presenta en la
infancia o adolescencia temprana.
La presentación más común
son
manifestaciones que
incluyen dolores óseos, alteración
en la
marcha, rigidez de
las articulaciones con dolor,
deformidades oseas visibles y fracturas patológicas. Puede afectar
a un
solo hueso denominándose displasia fibrosa
monostótica o numerosos huesos denominándose displasia fibrosa poliostótica. Aproximadamente el 70-80 %
de los casos posee una presentacion monostótica
y el 20-30 % son poliostóticos. En el esqueleto apendicular
por lo general se presenta
con
cojera y / o dolor, muchas veces los niños lo refieren como cansancio, en ocasiones una fractura patológica puede ocurrir como debut, cuando la
displasia fibrosa ocurre en los huesos craneofaciales generalmente se
presenta como un "bulto"
indoloro o asimetría facial, sin embargo, las áreas más comúnmente
involucrados son el fémur proximal y base del cráneo. (11) (5)
(12)
La
hiperpigmentación cutánea consiste
en manchas que van desde el marrón claro hasta
el
marrón oscuro, que a
menudo muestran una
distribución segmentaria, y con frecuencia predominan en un lado
del
cuerpo, pueden cruzar la línea media, pero
más
a menudo, respeta la línea media. Los lugares frecuentes son la nuca del cuello y el pliegue en el ápice
de las nalgas.
En retrospectiva, puntos de café con leche generalmente están presentes al nacer o bien se
presentan poco después. Como tal, pueden ser una pista temprana para el diagnóstico. Se han
descrito clásicamente como que
tienen una frontera
de "costa de Maine", que se refiere a la apariencia
irregular de la costa de Maine como aparece en los mapas, sin embargo, esto no siempre
se cumple. (2) (5) (7)
La
pubertad precoz
independiente de los niveles de
gonadotropinas usualmente
se manifiesta en las
niñas con sangrado o manchado vaginal,
acompañado por desarrollo de tejido mamario, generalmente sin desarrollo de vello púbico. Se
observan formas de
precocidad sexual en
más del
50% de las mujeres con SMA.
En los niños,
puede presentarse con
un agradamiento testicular bilateral o unilateral con agrandamiento del pene, rugosidad escrotal, olor corporal, vello púbico y axilar sumado a un
comportamiento sexual precoz. (2)
(5) (7)
El hipertiroidismo es
poco frecuente sin otras características del SMA también presentes,
los hallazgos pueden incluir taquicardia, arritmias, principalmente supra ventriculares,
hipertensión, hipertermia, temblor, insomnio, pérdida
de peso o retraso en el desarrollo en lactantes. Otras posibles manifestaciones que pueden coexistir incluyen al síndrome de Cushing, Exceso de hormona del crecimiento manifestado como gigantismo y acromegalia, tumores de
tiroides, tumores pituitarios, hipofosfatemia manifestado como
raquitismo hipofosfatémico, hipogonadismo hipogonadotrópico, particularmente en el contexto de hiperprolactinemia y
quistes en los ovarios (2) (11)
El SMA se puede confundir con la neurofibromatosis generalmente cuando un niño
presenta manchas cafés con leche. La
ubicación y la forma de
los puntos generalmente pueden ayudar a
distinguir el SMA de la
neurofibromatosis, ya que la
hiperpigmentación de SMA
tiene
bordes dentados, mientras que
la hiperpigmentación de la neurofibromatosis tiene
bordes suaves, pero
se debe considerar
un diagnóstico de neurofibromatosis si se observa un
historial familiar de pigmentación
de café con leche. En el SMA,
la enfermedad esquelética casi siempre implica uno
o ambos fémures proximales y / o
el
cráneo base, así como otros lugares, mientras que la
implicación esquelética en la neurofibromatosis
es poco común y generalmente involucra
las diáfisis de los huesos largos,
especialmente las tibias, a menudo
conducen a pseudoartrosis.
Cuando la pubertad precoz es el signo que se presenta en primera instancia, el diagnóstico
diferencial incluye
neoplasia
ovárica y pubertad precoz central, las pacientes con hemorragia
vaginal precoz o desarrollo
mamario, se deben considerar otras posibles causas de exceso de
estrógenos. La ingestión accidental de suplementos de
estrógenos puede
causar desarrollo de
los senos, aumento de la velocidad de la altura
y maduración del revestimiento endometrial. A
medida que los niveles de estrógeno disminuyen, puede producirse
un sangrado por deprivación Si se produce sangrado vaginal en ausencia de otros signos de exceso de
estrógenos como el desarrollo de
los senos o aumento
de la velocidad de
crecimiento, se debe
obtener una historia cuidadosa teniendo en cuenta posibles traumas o abusos sexuales. La Displasia osteofibrosa se
puede confundir con la displasia fibrosa del SMA, las lesiones de la displasia osteofibrosa
se suelen encontrar casi exclusivamente en la tibia y el peroné, y son histológicamente distintos de
la Displasia fibrosa. Por otro lado, El fibroma
no osificante puede compartir
similitudes
radiológicas es histológicas con la Displasia Fibrosa en los huesos largos. Es recomendable realizar un diagnóstico diferencial con diversas afecciones como: Displasia fibrosa monostótica, Neurofibromatosis, Síndrome de Cushing, Hipertiroidismo, Terapia con hormonas tiroideas, Terapia con glucocorticoides, Pubertad precoz de tipo central, Gigantismo y acromegalia, Tumor funcionante de ovario, Raquitismo hipofosfatémico, , Síndrome de Proteus, Síndrome
de Mazabraud y Enfermedad
de Paget. (2)
Se debe realizar estudios endocrinos
completos, los estudios de laboratorio que pueden
ser útiles incluyen los siguientes: Determinación de los niveles de
Gonadotropinas y niveles de
hormonas sexuales, biometría hemática completa y
química urinaria, prueba de tiroides
midiendo los niveles la hormona estimulante de la tiroides (TSH), tiroxina (T 4), anticuerpos antitiroideos, Niveles de hormona adrenocorticotropa
(ACTH), Prolactina sérica (PRL), Prueba de supresión con dexametasona (dosis estándar o
baja
/ dosis alta), recolección de orina analizada para
cortisol libre, niveles de la hormona del crecimiento (GH) y del factor de crecimiento similar a la insulina 1 (IGF-1) y prueba
de reacción en cadena de la
polimerasa (PCR), niveles de
calcio y fósforo séricos,
Fosfatasa alcalina sérica, N-telopéptidos e
hidroxiprolina urinarios,
Osteocalcina sérica
y enzimas hepáticas. Las modalidades de diagnóstico por imágenes que
se pueden considerar
incluyen
las
siguientes:
Radiografía
simple principalmente
cráneo
y
mandíbula, es decir un examen craneofacial,
pelvis, fémures, según lo indicado clínicamente, Ultrasonografía (cuello y tiroides, ovárico y pélvico, según lo indicado clínicamente
ya que permite detectar
y medir quistes de ovarios, Tomografía computarizada (TC)
que puede ser útil para detectar hiperplasia suprarrenal y atrapamiento de nervios intracraneales, Resonancia magnética (MRI); como clínicamente pertinente Escaneo
de radionucleidos
óseos (11) (5) (3) (8)
Con respecto a la histología, las areas afectadas por la displasia fibrosa se
componen por gran cantidad de
células parecidas a los fibroblastos con mínima matriz extracelular, abundancia de
células proosteogénicas que maduran en osteoblastos anormales. Existe un patrón óseo con aspecto desorganizado, semejante a una
"sopa alfabética" o de "letras chinas", el osteoide es de
forma irregular
(retorcido) con
un estroma fibroso muy celular. (2)
No
existe un tratamiento
específico para el SMA. Los agentes farmacológicos que
se han usado para tratar la pubertad precoz
en
SMA incluyen los siguientes Inhibidores de aromatasa,
Análogos de la hormona
liberadora de gonadotropina, Ketoconazol, Agonistas del receptor de
estrógeno como el tamoxifeno, Espironolactona,
Acetato de ciproterona, Acetato de medroxiprogesterona.
Actualmente, no hay
terapias médicas
clínicamente probadas
disponibles para la displasia osteofibrosa poliostotica
asociados con SMA. Los bisfosfonatos orales e intravenosos como el Pamidronato, alendronato, zoledronato, ayudan a aliviar el dolor y pueden ser beneficiosos para prevenir la progresión de la enfermedad, aunque los datos son
contradictorios. Los agentes farmacológicos que se han usado para tratar el hipertiroidismo en MAS incluyen los siguientes Tionamidas como el Propiltiouracilo, Metimazol, Radioyodo para la ablación de tejidos. No hay disponible
un tratamiento médico efectivo a
largo plazo para el
síndrome de Cushing independiente de ACTH. Los agentes farmacológicos que se han usado para
tratar el exceso de GH en SMA incluyen al octreotide, agonistas de la dopamina como la
Bromocriptina y la cabergolina, generalmente
junto con octreotida, antagonistas del receptor de
GH como el Pegvisomant probablemente no se usen mejor como monoterapia en este entorno.
(11) (4) (8)
El tratamiento farmacológico de
otras manifestaciones de SMA es
el siguiente:
• Hipogonadismo:
terapia de reemplazo hormonal apropiada
• Hipofosfatemia
con hiperfosfaturia: reemplazo agresivo de
fósforo oral
• Raquitismo hipofosfatémico
- Terapia apropiada de calcitriol con la reposición de calcio
y fosfato
• Las intervenciones quirúrgicas
que se pueden
considerar incluyen
las siguientes:
• Pubertad precoz: ooforectomía o cistectomía ovárica,
cuando falla el tratamiento
médico
• Displasia fibrosa poliostotica: tracción o fijación para fracturas; para la mayoría de las
lesiones. No se justifica la remoción rutinaria de la displasia
fibrosa poliostotica
• Hipertiroidismo: tiroidectomía
parcial,
casi
total o total
• Síndrome de Cushing Infantil - Adrenalectomía
bilateral
Gigantismo o acromegalia: la extirpación quirúrgica de
la lesión central rara vez es
curativa y solo se considera si el tumor amenaza
la visión en consulta previa
con
un neurocirujano experimentado en
base de cráneo (5)
(7) (8)
Con respecto a
la expectativa de vida de los pacientes con SMA, este
es
relativamente normal, ya que no se asocia a un aumento significativo en la mortalidad, sin embargo el
pronóstico y las complicaciones
pueden variar dependiendo
de las manifestaciones del SMA; la displasia fibrosa puede
tener efectos graves, que incluyen fracturas patológicas, desfiguración
facial y problemas de visión y audición.
Las terapias actuales se enfocan en el tratamiento de las complicaciones de la displasia
fibrosa, en lugar
de evitar
que se desarrolle. Los estudios actuales que usan bisfosfonatos son prometedores, aunque
no está claro si los bisfosfonatos reducen significativamente la
morbilidad asociada
con
estas lesiones. La
pubertad temprana
no es una afección potencialmente
mortal y no parece conducir a problemas clínicos posteriores.
Es posible que existan complicaciones debido a manifestaciones endocrinológicas que
se puede asociar
al SMA así, el hipertiroidismo puede ocasionar alteraciones del desarrollo en la infancia
y osteoporosis la taquicardia
resultante
de un
hipertiroidismo severo puede complicar o desencadenar un evento cardíaco; si se asocia un síndrome de Cushing infantil puede
causar una falla grave de crecimiento,
tono muscular deficiente
e hipertensión. (13) (8)
Bibliografía.
|
1.
|
Garcés J, Munduteguy M, Romero C. Síndrome de McCune-Albrigth
Evaluación del Compromiso Craneofacial y
de Columna por Imágenes de Resonancia Magnética. Neurorradiología Caso clínico. 2011 Enero; 75(1).
|
|
2.
|
Riverón DLOM.
Enfermedad de McCune-Albright. Revista Cubana
de
Ortopedia
y
|
|
Traumatología. 2005 Junio.
|
|
3.
|
POMA A. Síndrome
de McCune-Albright: Evaluación
del Compromiso Craneofacial
por Imágenes de Resonancia
Magnética.
Anales de
la
Facultad
de
Medicina Universidad
Nacional Mayor de San Marcos. 1999; 60(3).
|
|
4.
|
Ballesteros S. SÍNDROME DE McCUNE-ALBRIGHT: REPORTE DE CASO Y
REVISIÓN DE LA LITERATURA.
Rev.Medica.Sanitas. 2015
Octubre; 18(4).
|
|
5.
|
Collins C, Dumitrescu E, Michael T. McCune-Albright
syndrome. Orphanet Journal of Rare
Diseases. 2008 May; 3(12).
|
|
6.
|
HAPPLE R.
The McCune-Albright syndrome: a lethal gene surviving by mosaicism. Clinical
Genetics. 1986; 29.
|
|
7.
|
Hernández L, Espinosa M, Méndez
V.
Síndrome
de
McCune-Albright: características clínicas en una población pediátrica y adulta. Revista de Endocrinología y Nutrición. 2012
Enero; 20(1).
|
|
8.
|
Gabriel I,
Uwaif M. Medscape. [Online].; 2017 [cited
2017
April 26.
Available from:
https://emedicine.medscape.com/article/127233-overview.
|
|
9.
|
Pitarch Bort
G, Laguna Argente C, Martín
González B. Síndrome de McCune-Albright. Casos clinicos Servicio de Dermatología. Hospital General Universitari de València. 2009;
37(3).
|
|
10.
|
Martínez-Hervás S ea. Síndrome de McCune-Albright:
otra forma de neoplasias endócrinas
múltiples. Notas clínicas Servicio de Endocrinología
y Nutrición. Hospital Clínico Universitario de Valencia Departamento
de Medicina. 2005; 52(4).
|
|
11.
|
Bhadada SK. Fibrous
dysplasia & McCune-Albright
syndrome: An experience from
a
tertiary care centre in north India.
INDIAN J MED RES. 2011 May.
|
|
12.
|
Sabatel Hernández G ea. Displasia ósea fibrosa en el contexto de un síndrome de McCune-
Albright. Notas clinicas. 2004; 23(3).
|
|
13.
|
Garibaldi
L, Chemaitilly W.
A.D.A.M Multimedia Encyclopedia. [Online].; 2016 [cited 2016
August 16. Available from:
http://pennstatehershey.adam.com/content.aspx?productId=117&pid=1&gid=001217.
|

RECONOCIMIENTO-NOCOMERCIAL-COMPARTIRGUAL
CC BY-NC-SA
ESTA LICENCIA PERMITE A OTROS ENTREMEZLCAR, AJUSTAR Y CONSTRUIR A PARTIR DE SU OBRA CON FINES NO COMERCIALES, SIEMPRE Y CUANDO LE RECONOZCAN LA AUTORÍA Y SUS NUEVAS CREACIONES ESTÉN BAJO UNA LICENCIA CON LOS MISMOS TÉRMINOS.