
DOI: 10.26820/recimundo/4.(4).octubre.2020.371-381
URL: http://recimundo.com/index.php/es/article/view/914
EDITORIAL: Saberes del Conocimiento
Revista: RECIMUNDO
ISSN: 2588-073X
Tipo de Investigación: Artículo de Revisión
Código UNESCO: Ciencias Médicas
Paginas: 371-381
Noemi Andrea Barrera Zambrano1; Carlos Anthony Cárdenas Choez2; Juan Carlos Pincay Mendoza3; Ronald Adrián Valencia Rodríguez4
 https://orcid.org/0000-0002-7333-5149 https://orcid.org/0000-0002-1386-7741 https://orcid.org/0000-0002-2458-531X https://orcid.org/0000-0003-0730-3701
https://orcid.org/0000-0002-7333-5149 https://orcid.org/0000-0002-1386-7741 https://orcid.org/0000-0002-2458-531X https://orcid.org/0000-0003-0730-3701CORRESPONDENCIA
Noemi Andrea Barrera Zambrano
noemi_barrera15@hotmail.com
Guayaquil, Ecuador
La enfermedad de Behçet (EB) hace referencia a un desorden multisistémico de causas aún no bien conocidas, aunque algunos expertos han sugerido causas inmunitarias (y autoinmunitaria), virales o bacterianas e inclusive genéticas. Se caracteriza por úlceras orales y genitales recurrentes, uveítis, manifestaciones mucocutáneas, articulares, neurológicas, vasculares, intestinales y pulmonares. El objetivo perseguido es el de referir los criterios diagnósticos de la EB, en base a la literatura cientificoacadémica vigente, por ello, se escogió desarrollar una investigación de diseño bibliográfico, en el marco de una metodología de revisión. En los resultados se mostraron algunas acepciones vigentes y descripción de síntomas (signos), causas (etiología) y criterios diagnósticos de la EB. En conclusión, la EB es una patología crónica, incurable y difícil de diagnosticar. Además, también es posible atreverse a asegurar que, si bien los criterios del ISG aún son útiles, en la práctica médica actual cada vez más se usan los ICBD.
Palabras claves: Mucocutáneo, inmunológicos, HLA, autoinmunitaria, genéticas.
Behçet's disease (BE) refers to a multisystemic disorder whose causes are not yet well known, although some experts have suggested immune (and autoimmune), viral or bacterial, and even genetic causes. It is characterized by recurrent oral and genital ulcers, uveitis, mucocutaneous, articular, neurological, vascular, intestinal and pulmonary manifestations. The objective pursued is to refer to the diagnostic criteria of BE, based on the current scientific and academic literature, therefore, it was chosen to develop a bibliographic design research, within the framework of a review methodology. The results showed some current meanings and description of symptoms (signs), causes (etiology) and diagnostic criteria of BE. In conclusion, EB is a chronic, incurable and difficult to diagnose disease. Furthermore, it is also possible to dare to assure that, although the ISG criteria are still useful, ICBDs are increasingly used in current medical practice.
Keywords: Mucocutaneous, immunological, HLA, autoimmune, genetic.
A doença de Behçet (BE) se refere a um distúrbio multissistêmico cujas causas ainda não são bem conhecidas, embora alguns especialistas tenham sugerido causas imunológicas (e autoimunes), virais ou bacterianas e até genéticas. É caracterizada por úlceras orais e genitais recorrentes, uveítes, manifestações mucocutâneas, articulares, neurológicas, vasculares, intestinais e pulmonares. O objetivo prosseguido é fazer referência aos critérios diagnósticos da BE, com base na literatura científica e acadêmica atual, portanto, optou-se por desenvolver uma pesquisa de projeto bibliográfico, no âmbito de uma metodologia de revisão. Os resultados evidenciaram alguns significados e descrições atuais dos sintomas (sinais), causas (etiologia) e critérios diagnósticos de EB. Concluindo, a EB é uma doença crônica, incurável e de difícil diagnóstico. Além disso, também é possível ousar afirmar que, embora os critérios do ISG ainda sejam úteis, os ICBDs são cada vez mais utilizados na prática médica atual.
Palavras-chave: Mucocutâneo, imunológico, HLA, autoimune, genético.
INTRODUCCIÓN
El Instituto Nacional de Artritis y Enfermedades Musculoesqueléticas y de la Piel de los EEUU (NIAMS, por sus siglas en inglés) establece que la enfermedad de Behçet (EB) consiste en una afección crónica que se caracteriza por la aparición de ciertos signos o síntomas comunes, tales como: aftas (llagas) bucales, genitales y en otras áreas del cuerpo (mayormente en las piernas y en el torso superior), inflamación en la parte media o posterior del ojo (uveítis) y artritis (inflamación, dolor y rigidez en las articulaciones). En ningún caso se trata de un síndrome contagioso. ya que no se transmite de una persona a otra. En otros casos más raros y severos de esta enfermedad, se incluyen otros síntomas, tales como: coágulos sanguíneos (comúnmente en las piernas), inflamación del tracto digestivo y de la membrana que recubre el cerebro o médula espinal. (NIAMS, 2015)
Los médicos no están seguros de cuál es la causa del síndrome de Behcet. Es poco frecuente en Estados Unidos, pero es común en el Medio Oriente y Asia. Afecta principalmente a las personas entre los 20 y los 30 años de edad. El diagnóstico puede tomarse mucho tiempo, ya que los síntomas pueden aparecer y desaparecer y quizá pueden pasar meses o años hasta que aparezcan todos los síntomas.
No existe una cura. El tratamiento se enfoca en la disminución del dolor y la prevención de los problemas más graves. La mayoría de las personas pueden controlar los síntomas con el tratamiento. (MedlinePlus, 2019)
La EB tiene un patrón geográfico único con una inusual y alta incidencia en a lo largo de la conocida “ruta de la seda”, que abarca lo que son países del mediterráneo, Del Medio Oeste y lejano Oeste. (Maldonado, 2012) en Turquía en >1/1.000, frente a 1/10.000 en Japón. Los casos europeos se describen con mayor frecuencia en los países mediterráneos. (Consorcio Orphanet, 2020) En la mayoría de las series, no hay diferencias en cuanto al género en los niños, en contraste con la EB en los adultos, en donde los hombres con dos veces más afectados que las mujeres. En promedio, 5.4% a 6.7% de todos los pacientes tiene inicio de la EB en la infancia. La alta frecuencia en Japón y países de medio oeste y la alta frecuencia de ocurrencia familiar sugieren componente genético o ambiental en la enfermedad. EL HLA B51 ha sido fuertemente asociado con este síndrome en la población adulta. (Maldonado, 2012)
Esta enfermedad se presenta en ambos sexos, aunque tiende a ser más grave en hombres, y suele comenzar en la tercera década de la vida. A veces puede aparecer en niños. La incidencia varía según la ubicación geográfica.
La causa de la enfermedad de Behçet se desconoce. Se han sugerido causas inmunitarias (incluyendo una etiología autoinmunitaria) y virales o bacterianas, y HLA-B51 es un factor de riesgo mayor. La prevalencia de un alelo HLA-B51 es > 15% entre las personas de Europa, Oriente Medio y el Lejano Oriente, pero es baja o ausente entre las personas procedentes de África, Oceanía y América del Sur. (Villa, 2019)
Vargas, Dávila, Puerres, Álvarez, & Capelo (2019) señalan que:
La EB pertenece al conjunto de las consideradas autoinmunes y autoinflamatorias. Puede afectar a cualquier vaso sanguíneo del cuerpo, pero no a todos los casos por igual. Acostumbra a manifestarse primeramente en la etapa juvenil, cuando el individuo se encuentra en plena formación y programación de su futuro. Resulta dolorosa, invisible, tanto en las analíticas como socialmente, crónica y en alto grado invalidante. Tarda una media de 5 a 10 años en ser diagnosticada y resulta ineludible que los enfermos, tanto antes como después del diagnóstico, padezcan soledad e incomprensión.
En la presente entrega el objetivo perseguido es el de referirlos criterios diagnósticos de la EB, en base a la literatura cientificoacadémica vigente. A continuación, se describe la metodología empleada, y en los resultados se muestran algunos aportes respectos a concepciones, síntomas (signos), causas (etiología) y criterios diagnósticos de la EB.
Materiales y Métodos
Con la finalidad de realizar el presente estudio se llevó a cabo una búsqueda para recolectar y seleccionar el material bibliográfico en formato digital, que fue la base para analizar y sintetizar el tema de: Criterios de enfermedad de Behçet, por lo cual, la presente investigación se define dentro de un diseño documental o bibliográfico y de una metodología de revisión.
Fueron usadas varias bases de datos y páginas web relacionadas con el área de la salud de carácter nacional e internacional, con validez científica y reconocida, para ubicar el material que sirvió de fundamento cientificoacadémico en ésta entrega. Algunas de esas bases de datos fueron: BVS PubMed, SciELO, Base, Medigraphic, Redalyc; y entre los sitios web consultados figuraron: MedlinePlus, Manuales MSD, y otras. El tipo de material bibliográfico que se consideró escoger debía estar concebido como: artículo científico original. tesis de grado, postgrado o doctorado, reporte de caso clínico, guía clínica, protocolo, consenso, informe académico de práctica profesional, y otras clases de contenidos que se sustentaran en cualquier clase de evidencia recogida bajo métodos científicos.
Los contenidos repetidos fueron desestimados, así como editoriales o cartas editoriales, anotaciones académicas y cualquier otro tipo de material bibliográfico carente de fuentes de sustento científico o con bajo nivel de evidencia. La búsqueda fue llevada a cabo durante el mes de octubre del presente año, utilizando palabras clave y operadores lógicos (o booleanos) que, a manera de ecuaciones de búsqueda se fueron configurando, por ejemplo: “criterios + enfermedad | síndrome + Behçet” y “criteria + disease | syndrome + Behçet”.
Los registros bibliográficos ubicados fueron filtrados, principalmente, en base a los criterios de: idioma español; relevancia del tema y/o mayor correlación temática posible; fecha de publicación de los últimos 10 años; no obstante, otros tipos de variables de refinamiento de resultados fueron igualmente aplicados según estuvieran disponibles de manera particular en cada base de datos.
Por último, se considera importante aclarar que tanto la metodología como el análisis y la argumentación que aquí se expone se efectuó de manera consensuada entre los miembros del equipo investigador.
Resultados
La enfermedad de Behçet (EB) es un desorden multisistémico de etiología desconocida caracterizado por úlceras orales y genitales recurrentes, uveítis, manifestaciones mucocutáneas, articulares, neurológicas, vasculares, intestinales y pulmonares. (Maldonado, 2012)
Para el Consorcio Orphanet (2020) se trata de “una vasculitis multisistémica, crónica y reincidente, caracterizada por lesiones mucocutáneas, así como por manifestaciones articulares, vasculares, oculares y del sistema nervioso central”. En MedlinePlus (2019) se aclara que “Los problemas más serios pueden incluir meningitis, coágulos sanguíneos, inflamación del sistema digestivo y ceguera” .
El inicio ocurre con mayor frecuencia en adultos (media de 30 años de edad), pero se han notificado casos pediátricos. Los episodios recurrentes de afrathas orales redondas con bordes eritematosos agudos y elevados (1-3 cm de diámetro) pueden ir acompañados de aphthae genital (>50% de los casos); Las características cutáneas pueden incluir pseudo-foliculitis y eritema nodoso. Los trastornos oculares (uveítis posterior, vasculitis de la retina) ocurren en más del 50% de los pacientes con BD. Artralgia y/o no erosivo, asimétrico, artritis que afecta principalmente a la articulación grande (rodillas, tobillos ect.) son frecuentes (45%) y puede ocurrir como un síntoma inicial. La vasculitis en BD es más frecuente en el sistema venoso donde pueden ocurrir trombosis en territorios femoro-ilíacos, superiores e inferiores de vena cava y cerebrales. Las trombosis arteriales y aneurismas raros afectan principalmente a los vasos pulmonares y aorta. Las manifestaciones neurológicas (neuro-BD) son frecuentes (>20%), y pueden incluir dolor de cabeza, fiebre, signos piramidales con hemiparesia, daño craneal al nervio, meningitis, cambios de comportamiento y disfunción del esfínter. Las lesiones aphtoide y/o ulcerosas pueden afectar todo el tracto digestivo, pero principalmente el ileo-caecum y el colon ascendente, lo que puede provocar hemorragias y perforaciones. (Consorcio Orphanet, 2020)
Síntomas (Signos)
Usualmente, la fiebre y el malestar general son síntomas que pueden ocurrir en la EB, sin embargo, los más característicos son los que se manifiestan en el tejido mucocutáneo (ulceraciones bucales, genitales, tronco y extremidades) y los oculares (inflamación); cuando se presenta de forma más severa, los signos alcanzan las lesiones vasculares, gastrointestinales y neurológicas. (Villa, 2019)
En Gómez, Vera & Martínez (2017) se ha encontrado un caso típico que bien sirve de ejemplo para ilustrar mejor la idea (ver Figura 1) antes expuesta, ya que los mismos describieron que en su exploración clínica pudieron detectar úlceras cervicovaginales (cuadrantes gráfico “A” y “B”). En el tronco presentaba pequeñas pústulas (cuadrante gráfico “C”). En la lengua y la mucosa yugal también se detectaron numerosas úlceras (cuadrante gráfico “D”). El caso fue de:
Una mujer de 19 años con una historia previa de estomatitis aftosa desarrolló múltiples nódulos eritematosos y dolorosos en los miembros inferiores, acompañados de fiebre y dolor vulvar. Acudió a Urgencias y tras la inyección de diclofenaco intramuscular presentó rápidamente una reacción local en la zona de la inyección con pequeñas vesículas.
En base a (Campos, Baixauli, Rueda, & Calvo, 2013) Villa (2019) es posible detallar que, en las usuales y dolorosas (en ciertos casos) lesiones mucocutáneas, las úlceras (bucales y/o genitales) están generalmente entre los primeros signos y síntomas. Las características de éstas es que pueden presentarse de forma redonda u ovalada, llanas o agudas, con un diámetro de 2 a 10 mm y un área central necrótica amarillenta. Se detectan en el interior de la cavidad bucal, comúnmente agrupadas, y en promedio se mantienen entre 7 y 14 días. Igualmente pueden aparecer en la vulva, el pene y/o el escroto, sitios donde suelen ser dolorosas, pero las que salen en la vagina, tienden a ser indoloras.
Las afecciones oculares oscilan entre el 25 y 75% de los pacientes con EB, pudiendo estar vinculadas con síntomas neurológicos. Estos daños pueden tratarse de: uveítis (bilateral y episódica) o iridociclitis recidivante, hipopión, panuveítis; y en situaciones más delicadas, coroiditis, vasculitis retiniana, oclusión vascular y neutiris óptica, condiciones que pueden afectar irreversiblemente la visión, al progresar inclusive a la ceguera.
A nivel musculoesquelético, los signos que medianamente se presentan pueden ir desde artralgias (autolimitadas, no destructivas y parcialmente leves) hasta la artritis propiamente, particularmente en las rótulas y otras articulaciones importantes. Otras manifestaciones incluyen: molestias abdominales, diarrea y dolor (a raíz de úlceras en el colon o íleon) e inflamación sacroilíaca
Las complicaciones vasculares son múltiples y serias. La inflamación en las venas y arterias puede avanzar a nivel peri y endovascular. En las arterias podría sobrevenirse una trombosis (en venas superficiales y profundas), aneurisma (graves en la arteria aorta y pulmonar), seudoaneurisma; estando éstas entre las más frecuentes. También es posible que ocurra una hemorragia, estenosis una hemoptisis (a raíz de fístulas entre la arteria pulmonar y un bronquio). Aunque las afecciones de grandes arterias causadas por esta patología podrían consistir de un 3 al 5% a lo largo de su vida, con las autopsias de los mismos se han evidenciado daños de grandes vasos asintomáticos hasta en un 33%. Es posible que más de una vena pueda verse afectada, inclusive, la vena cava inferior y superior, las venas hepáticas (que causan el síndrome de Budd-Chiari), y los senos venosos durales.
La afección del sistema nervioso central es menos frecuente, aunque grave. Puede ser de comienzo brusco o gradual. Las primeras manifestaciones pueden ser la afección del parénquima con signos piramidales, afección de pequeños vasos con patrón de esclerosis múltiple, falta de compromiso del parénquima con meningitis o meningoencefalitis aséptica o trombosis de senos durales. La meningitis aséptica, en el contexto clínico característico, puede sugerir el diagnóstico.
Luego de varios años, pueden aparecer afecciones psiquiátricas con cambios de personalidad y demencia. La neuropatía periférica, frecuente en otras afecciones vasculíticas, es inusual en la enfermedad de Behçet. (Villa, 2019)
Etiología
De origen desconocido, la predisposición genética en EB puede permitir que ciertos insultos infecciosos (en particular Streptococcus sanguis) y/o ambientales desencadenen síntomas que impliquen ataques inflamatorios esporádicos que recuerden a trastornos autoinflamatorios debido a reacciones cruzadas con antígenos de mucosa oral. El antígeno HLAB5101 se asocia a la EB en el 50-60% de los pacientes. La activación de NF-kB y los niveles aberrantes de citoquinas (por ejemplo, IL-6, TNF-a, IL-8, IL-12, IL-17 e IL-21) han sido implicados en la patogénesis de BD. Una forma autosómica dominante familiar de BD, haploinsuficiencia A20, está relacionada con mutaciones en TNFAIP3(6q23.3). (Consorcio Orphanet, 2020)
Aunque la etiología del SB es desconocida, existen numerosas investigaciones que señalan la participación de factores genéticos, infecciosos (virales y bacterianos) e inmunológicos. Entre los más importantes se señalan la asociación con el genotipo HLA de los pacientes, la reactividad cruzada con péptidos humanos y la activación del endotelio vascular. (Alfonso, 2016, pág. 303)
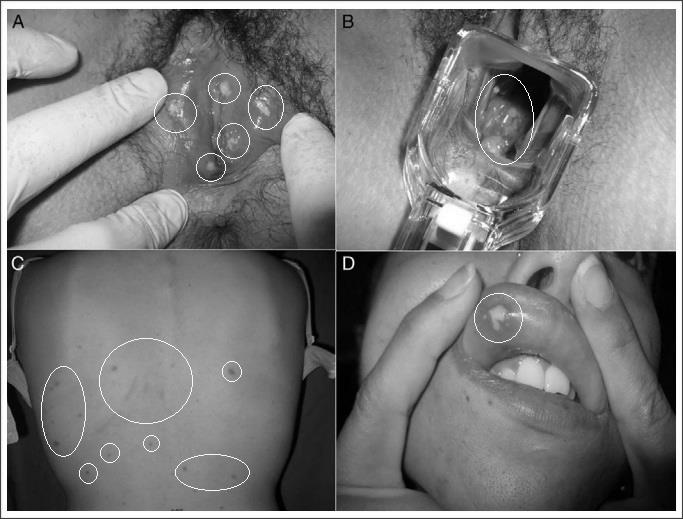
Figura 1. Caso típico de manifestaciones clínicas de EB en mujer.
Fuente: (Alfonso, 2016, pág. 303)
La EB también puede aparecer en la infancia, en general con formas incompletas. Las manifestaciones más comunes son las úlceras orales y genitales, lesiones cutáneas, artritis y uveítis. Algunos recién nacidos de mujeres con EB presentan úlceras orales y genitales, pústulas y úlceras necróticas cutáneas de distribución periungueal. Estas lesiones son autolimitadas y desaparecen en seis semanas. El curso de la RB durante el embarazo es variable. Puede producirse activación, remisión o cambios clínicos en cada caso. (Castillo, González, & Hernández, 2014, pág. 315)
Criterios Diagnósticos
Son mayores las coincidencias respecto a lo difícil que es diagnosticar de ésta patología (Taroco, Fernández, Maciel, Consani, & Facal, 2009) por cuanto, no hay una prueba o examen específico para ello, sus síntomas, en muchos casos, no suelen evidenciarse de manera clara o pueden retardar su aparición incluso luego de varios años (NIAMS, 2015; MedlinePlus, 2019); de allí la importancia del diagnóstico diferencial, el cual podría tratarse de: enfermedad de Crohn, artritis reactiva, ulceraciones por herpes, artropatías espondilo, uveítis infecciosa, policondritis recidivante, sarcoidosis, síndrome antifosfolípido, arteritis de Takayasu, meningoencefalitis infecciosa o esclerosis múltiple. (NIAMS, 2015; Vargas et al., 2019; Consorcio Orphanet, 2020). Además, no hay resultados de laboratorio patognomónicos asociados a su determinación específica. Es la clínica, entonces, lo que termina siento la base fundamental de su diagnosis, así lo dejan ver la mayoría de las fuentes consultadas. (Taroco et al., 2009; Camposet al., 2013)
Otras características que sugieren un considerable incremento en la probabilidad de su diagnóstico también puede estar representado por los antecedentes familiares de EB, la afectación visceral grave aislada (indistintamente de que se manifieste inicialmente) y, en casos de imprecisión diagnóstica, es bastante valiosa la determinación del haplotipo HLA-B51; éste último, según Gutiérrez et al. (2018), asociado en países orientales “con formas más severas de afectación ocular” (pág. 132) y en los occidentales con el HLA-B57; aunque por sí solo no se le considere un poderoso elemento de diagnóstico. (Consorcio Orphanet, 2020)
En principio, la mayoría de los estudios consultados, aparte de convalidar en gran medida las ideas que en general se han señalado en cuanto al diagnóstico, también concuerdan en referir como guía para tales fines, los propuestos por el grupo internacional de estudio (ISG, del inglés International Study Group) de la enfermedad de Behçet (Maldonado, 2012; Campos et al.; 2013; Alfonso, 2016; Gutiérrez et al., 2018)
Alfonso (2016), de manera detallada explica que la definición de dichos criterios clínicos fueron se basan en las manifestaciones de los siguientes signos en un paciente:
Seguidamente sustenta que:
Aunque validados, los criterios diagnósticos del grupo internacional de estudio de la enfermedad de Behcet no pueden sustituir la valoración clínica individual de los casos. El 3 % de los enfermos no presentan úlceras orales y, por otra parte[sic] las múltiples manifestaciones de la enfermedad no se presentan todas al inicio de la enfermedad y algunas pueden tardar años en aparecer. (pág. 307)
Se considera oportuno y valioso lo señalado en Castillo, González, & Hernández (2014) ya que éstos hacen el recordatorio de que dichos criterios fueron establecidos por primera vez en 1990, y luego fueron se revisaron en 2006 (ver Tabla 1), oportunidad en la que la propuesta incluyó otros criterios, tales como: las trombosis arteriales, venosas, y aneurismas de grandes vasos, con los que se obtenía mayor especificidad diagnóstica. (pág. 310)
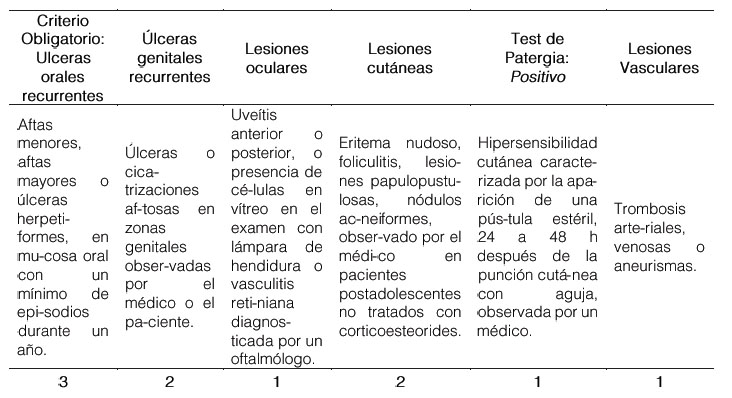
Tabla 1. Criterios diagnósticos del Grupo Internacional para el estudio (ISG) de la enfermedad de Behçet (Revisión 2006).
Nota: Adaptado de “Enfermedad de Behçet” Castillo et al. (2014). Revista Cubana de Reumatología. 16(3). Pág. 310. Recuperado de: https://www.researchgate.net/publication/281594241_Enfermedad_de_Behcet. Cada columna hace referencia al criterio considerable para EB, su descripción y el valor (puntaje) que representa para determinar su ocurrencia; verificable por la sumatoria de los valores de las concurrencias (incluida la obligatoria) si es mayor o igual a 4 puntos.
En el 2013, un grupo internacional, compuesto por representantes de 27 países, revisaron los criterios internacionales para el diagnóstico de la enfermedad de Behçet (ICBD, por sus siglas en inglés), analizaron datos de 2556 pacientes diagnosticados clínicamente y 1 163 controles con enfermedades que simulan el Behcet o que presenten al menos un signo mayor del síndrome. Como resultado del análisis propusieron otorgar 2 puntos a las lesiones oculares y a las lesiones aftosas orales y genitales y asignar 1 punto a las lesiones de piel, del SNC, las manifestaciones vasculares y al test de patergia positivo. De acuerdo a estos criterios, un paciente con una puntuación ≥ 4 puntos se clasifica como una enfermedad de Behçet. Estos nuevos criterios tienen una mayor sensibilidad que los del grupo internacional de estudio (94,8 % vs 85 %) y mantienen una especificidad aceptable, pero menor (90,5 % vs 96 %) (Alfonso, 2016, pág. 307)
No obstante, en otros recursos bibliográficos publicados más recientemente (Campos et al.; 2013; Álvarez et al., 2020; Consorcio Orphanet, 2020) se destacan los Criterios Internacionales para Enfermedad de Behçet (ICBD, del inglés International Criteria for Behçet's Disease) como la base del diagnóstico de la EB.
De hecho, Álvarez et al. (2020), dejan en evidencia un estudio comparativo retrospectivo entre los criterios del ISG y los ICBD en el diagnóstico de EB, en base a 111 pacientes españoles, los cuales fueron bien definidos y diagnosticados entre 1980 y 2019 de EB posible o definitiva por parte de reumatólogos expertos, señalaron que, si bien los criterios del ISG venían siendo la clasificación diagnóstica más utilizada, los mismos han resultado ser poco sensibles, siendo por ello que en 2014 se originan los ICBD.
Algunos de los datos más destacables en sus resultados se refieren a: el diagnóstico efectivo de dicha patología en 65 pacientes mediante los criterios ISG, mientras que con los ICBD se determinar en 86 de los mismos. Entre esos dos grupos no se hallaron diferencias estadísticamente significativas. Se pudo determinar una moderada concordancia entre ambos criterios diagnósticos. En definitiva, concluyeron que “Los criterios ICBD han demostrado una sensibilidad más alta que los ISG. Por ello, la aplicación de los nuevos criterios puede suponer un diagnóstico más temprano y correcto de la EB.” (Álvarez et al., 2020, pág. 296)
Otra fuente consistente con la idea antes referida es representada por el Consorcio Orphanet (2020) ya que éstos explican que la diagnosis de la EB es fundamentalmente clínico y en razón de la obtención de 4 o más puntos tras la aplicación de los ICBD, derivados de la ponderación clasificada de la siguiente manera: ulceración oral recurrente (al menos 3 veces durante 12 meses; 2 puntos), ulceración genital (2 puntos), uveítis (2 puntos), lesiones cutáneas (1 punto), cardiovascular (1 punto), neurológica (1 punto) y/o reacción patérgica (1 punto). No obstante, el diagnóstico también es posible al considerar otros elementos contributivos, tales como: la afectación visceral grave aislada (trombosis venosa cerebral y/o subhepática; afectación neurológica y/o vasculitis retiniana; vena cava; aneurismas pulmonares) independientemente de su eventualidad inicial; antecedentes familiares de EB; el hallazgo del haplottipo HLA-B51 (muy útil en situaciones de incertidumbre diagnóstica); por lo tanto, en todo momento es valiosa la opinión del médico especialista de forma sistemática.
Conclusión
Sobre la base de las fuentes de datos utilizadas en este estudio, ha de comprenderse que la EB es una patología crónica, incurable y difícil de diagnosticar. Además, también es posible atreverse a asegurar que, si bien los criterios del ISG aún son útiles, en la práctica médica actual cada vez más se usan los ICBD.
REFERENCIAS BIBLIOGRÁFICAS
Alfonso, M. (julio-septiembre de 2016). Síndrome de Behcet. Revista Cubana de Hematología, Inmunología y Hemoteraia, 32(3). Recuperado el 10 de octubre de 2020, de http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892016000300003
Álvarez, C., Herrero, A., Sánchez, L., Martínez, D., Martín, J., Suárez, G., . . . Blanco, R. (2020). Enfermedad de Behçet: Comparación de dos criterios diagnósticos en una población bien definida. Estudio de 111 pacientes. Reumatología Clínica, 16(Especial Congreso), 295-296. Recuperado el 10 de octubre de 2020, de https://www.reumatologiaclinica.org/es-congresos-xlvi-congreso-nacional-sociedad-espanola-111-sesion-posteres-6095-enfermedad-de-behcet-comparacion-de-72596-pdf
Campos, C., Baixauli, A., Rueda, A., & Calvo, J. (2013). Enfermedad de Behçet. En M. Belmonte, J. Castellano, J. Román, & J. Rosas, Enfermedades Reumáticas. Actualización SVR (II ed., Vol. Electrónico, pág. 1056 pp.). Valencia, España: Sociedad Valenciana de Reumatología. Recuperado el 10 de octubre de 2020, de https://svreumatologia.com/enfermedades-reumaticas-2/
Castillo, W., González, J., & Hernández, J. (enero de 2014). Enfermedad de Behçet. Revista Cubana de Reumatología, 16(3), 309-321. Recuperado el 10 de octubre de 2020, de https://www.researchgate.net/publication/281594241_Enfermedad_de_Behcet
Consorcio Orphanet. (Diciembre de 2020). Enfermedad de Behçet. Recuperado el 10 de octubre de 2020, de orpha.net: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=117&lng=ES
Gomez, E., Vera, A., & Martínez, L. (2017). Enfermedad de Behcet. Medicina Clínica, 151(4), 23. doi:10.1016/j.medcli.2017.11.051.
Gómez, E., Vera, A., & Martínez, L. (2017). Enfermedad de Behcet. Medicina Clínica, 151(4), 23. doi:10.1016/j.medcli.2017.11.051
Gutiérrez, I., Bulacio, E., Camporro, F., Bertorello, M., Furrer, S., Bressa, V., & Lucero, P. (2018). Úlceras orales a repetición y eritema nodoso: pensar en Enfermedad de Behçet. Revista Methodo: Investigación Aplicada a las Ciencias Biológicas, 3(4), 131-138. doi:10.22529/me.2018.3(4) 07
Maldonado, M. (enero de 2012). Guías de abordaje diagnóstico y terapéutico. (Algoritmo de 12 padecimientos). Recuperado el 10 de octubre de 2020, de himfg.edu.mx: http://himfg.com.mx/descargas/documentos/planeacion/guiasclinicasHIM/Greumatologia.pdf
MedlinePlus. (28 de junio de 2019). Síndrome de Behcet. Recuperado el 10 de octubre de 2020, de Medlineplus.gov: https://medlineplus.gov/spanish/behcetssyndrome.html
NIAMS. (01 de agosto de 2015). Síndrome de Behçet. Recuperado el 10 de octubre de 2020, de niams.nih.gov: https://www.niams.nih.gov/health-topics/behcets-disease/advanced#tab-overview
Taroco, R., Fernández, A., Maciel, G., Consani, S., & Facal, J. (octubre de 2009). Enfermedad de Behçet. Un diagnóstico elusivo. Tendencias, 35, 11-16. Recuperado el 10 de octubre de 2020, de http://clinicamedica1.com.uy/wp-content/uploads/2016/05/Enfermedad-de-Behcet.pdf
Vargas, A., Dávila, M., Puerres, D., Álvarez, M., & Capelo, T. (2019). Enfermedad de Behçet. Reflexiones sobre su diagnóstico y tratamiento. Revista Cubana de Reumatología, 21(3), 10 pp. Recuperado el 10 de octubre de 2020, de http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1817-59962019000300015
Villa, A. (marzo de 2019). Enfermedad de Behçet. Recuperado el 10 de octubre de 2020, de MDS Manuals [En Español]: https://www.msdmanuals.com/es/professional/trastornos-de-los-tejidos-musculoesquel%C3%A9tico-y-conectivo/vasculitis/enfermedad-de-beh%C3%A7et