
DOI: 10.26820/recimundo/4.(4).octubre.2020.392-398
URL: http://recimundo.com/index.php/es/article/view/916
EDITORIAL: Saberes del Conocimiento
Revista: RECIMUNDO
ISSN: 2588-073X
Tipo de Investigación: Artículo de Revisión
Código UNESCO: Ciencias Médicas
Paginas: 392-398
Cristhian Rubén Vallejo Zambrano1; Mendel Arnaldo Steinzappir Navia2; Simón Alfonso Ávila Meza3; María Cristina Azua Zambrano4; Karla Belén Zambrano Vásquez5; Miguel Eduardo Chumo Rivero6
 https://orcid.org/0000-0001-5513-8507 https://orcid.org/0000-0002-2183-9070 https://orcid.org/0000-0002-2242-6091 https://orcid.org/0000-0001-6700-4397 https://orcid.org/0000-0002-3537-1165 https://orcid.org/0000-0001-6789-0093
https://orcid.org/0000-0001-5513-8507 https://orcid.org/0000-0002-2183-9070 https://orcid.org/0000-0002-2242-6091 https://orcid.org/0000-0001-6700-4397 https://orcid.org/0000-0002-3537-1165 https://orcid.org/0000-0001-6789-0093CORRESPONDENCIA
Cristhian Rubén Vallejo Zambrano
mdcardiologycrvz@hotmail.com
Manta, Ecuador
Huntington es un desorden neurodegenerativo raro en nuestra sociedad, que afecta el sistema nervioso central (SNC) y se caracteriza por demencia, disturbios psiquiátricos, de comportamiento y movimientos involuntarios (corea de Huntington). Afecta en su mayoría a los pacientes de piel blanca y la edad aproximada de inicio está entre los 30-50 años; sin embargo, pueden darse casos en los que inicie a los 20 años (enfermedad de Huntington juvenil). El síndrome de Huntington (SH) es una enfermedad hereditaria autosómica dominante, causada por una elongación repetida del CAG en el cromosoma 4, en el gen Huntingtine. Para su diagnóstico, nos basamos en los síntomas y signos clínicos y lo confirmamos con una determinación de ADN. El diagnóstico prenatal es posible mediante una amniocentesis. Debemos tener en cuenta que, aunque no existe cura para esta enfermedad, si existen tratamientos que mejoren la calidad de vida de los pacientes afectados. El objetivo de este estudio es consolidar las teorías actuales asociadas al SH. Esta revisión fue elaborada mediante un análisis minucioso de artículos informáticos indexados en fuentes de información médica: Pubmed, Medline, Pubmed Central; siendo un estudio documental bibliográfico. Es de vital importancia, tener un estudio referente a esta enfermedad, en el cual se pueda entender de una manera precisa la fisiopatología; para posteriormente, dar un buen tratamiento sintomatológico a los pacientes con SH.
Palabras claves: Huntington; Autosómico; Determinación; Tratamiento.
Huntington is a rare neurodegenerative disorder in our society, which affects the central nervous system (CNS) and is characterized by dementia, psychiatric and behavioral disorders and involuntary movements (Huntington's chorea). It mostly affects patients with white skin and the approximate age of onset is between 30-50 years; however, there may be cases where it begins at age 20 (juvenile Huntington's disease). Huntington syndrome (HS) is an autosomal dominant inherited disease, caused by a repeated elongation of the CAG on chromosome 4, in the Huntingtine gene. For its diagnosis, we rely on clinical signs and symptoms and confirm it with a DNA determination. Prenatal diagnosis is possible by amniocentesis. We must bear in mind that, although there is no cure for this disease, there are treatments that improve the quality of life of affected patients. The objective of this study is to consolidate current theories associated with HS. This review was prepared through a careful analysis of indexed computer articles in medical information sources: Pubmed, Medline, Pubmed Central; being a bibliographic documentary study. It is vitally important to have a study regarding this disease, in which the pathophysiology can be understood in a precise way; to later, give a good symptomatological treatment to patients with HS.
Keywords: Huntington; Autosomal; Determination; Treatment.
Huntington é uma doença neurodegenerativa rara em nossa sociedade, que afeta o sistema nervoso central (SNC) e é caracterizada por demência, transtornos psiquiátricos e comportamentais e movimentos involuntários (coreia de Huntington). Afeta principalmente pacientes com pele branca e a idade aproximada de início é entre 30-50 anos; no entanto, pode haver casos em que começa aos 20 anos (doença de Huntington juvenil). A síndrome de Huntington (SH) é uma doença hereditária autossômica dominante, causada por um alongamento repetido do CAG no cromossomo 4, no gene Huntingtine. Para o seu diagnóstico, contamos com os sinais e sintomas clínicos e confirmamos com a determinação do DNA. O diagnóstico pré-natal é possível pela amniocentese. Devemos ter em mente que, embora não haja cura para esta doença, existem tratamentos que melhoram a qualidade de vida dos pacientes afetados. O objetivo deste estudo é consolidar as teorias atuais associadas ao HS. Esta revisão foi elaborada por meio de uma análise cuidadosa de artigos de computador indexados em fontes de informação médica: Pubmed, Medline, Pubmed Central; sendo um estudo bibliográfico documental. É de vital importância um estudo a respeito dessa doença, em que a fisiopatologia possa ser compreendida de forma precisa; para depois dar um bom tratamento sintomatológico aos pacientes com HS.
Palavras-chave: Huntington; Autosomal; Determinação; Tratamento.
INTRODUCCIÓN
Síndrome de Huntington
El Síndrome de Huntington (SH) es una rara y grave enfermedad neurológica, causada por un desorden neurodegenerativo hereditario, que se encuentra asociado con una afectación en el cromosoma 4, e involucra una repetición del trinucleotido CAG (citosina, adenina, guanina). Se caracteriza por presentar movimientos involuntarios de tipo hipercinético espontáneo, arrítmicos, excesivos y abruptos (Roos, 2010).
Este trastorno neurodegenerativo autosómico dominante, es también conocido como corea de Huntington y muchas veces como baile de San Vito¨ (en inglés Chorea Sancti Viti). Su ocurrencia se observó, bajo un contexto religioso, en la edad media, y asociado a demencia, disturbios psiquiátricos de comportamiento y movimientos involuntarios (Roos, 2010). Fue descrito por primera vez en 1872 por el médico George Huntington, cuando realizó una completa descripción clínica al estudiar la enfermedad dentro de una familia, en Long Island, desde la de los abuelos (Bruyn GW, 1968).
En la actualidad, existen muchos tratamientos sintomáticos, pero se sabe de avances farmacológicos para contrarrestar de una manera más adecuada este desorden, dependiendo de las especificidades de signos y síntomas clínicos. Por lo general, la hiperquiinesia o corea, es tratada con los bloqueantes de receptores de dopamina
En el presente estudio bibliográfico analizaremos de manera exhaustiva la epidemiologia, fisiopatología, síntomas y signos clínicos, diagnóstico y tratamiento asociado a esta enfermedad que avanza de manera crónica, y para la cual no existe cura en la ciencia actual.
Epidemiologia
El SH tiene una prevalencia de 5-10 pacientes caucásicos de 100.000, presentan la enfermedad (Bates G, Harper P, & Jones L, 2002). En Europa, esta enfermedad tiene una incidencia de 3 a 7 casos por cada 100.000 habitantes; mientras que en Estados Unidos y Chile es de 4 a 8 por cada 100.000 habitantes (Conneally, 1984).
Fisiopatología:
Un paciente con uno de los padres con SH se divide en un riesgo por etapas: preclínico (A) y clínico (B) (Roos, 2010). El estadio preclínico se subdivide en tres etapas: etapa de riesgo (50%), etapa de portador de gen y, por último, la etapa de transición.
El curso clínico se divide en tres etapas. La etapa uno, es el inicio de los síntomas, en la etapa dos se presentan disturbios motores más generalizados y en la etapa tres se caracteriza por disturbios motores severamente generalizados (Tabla 1). (Roos, 2010)
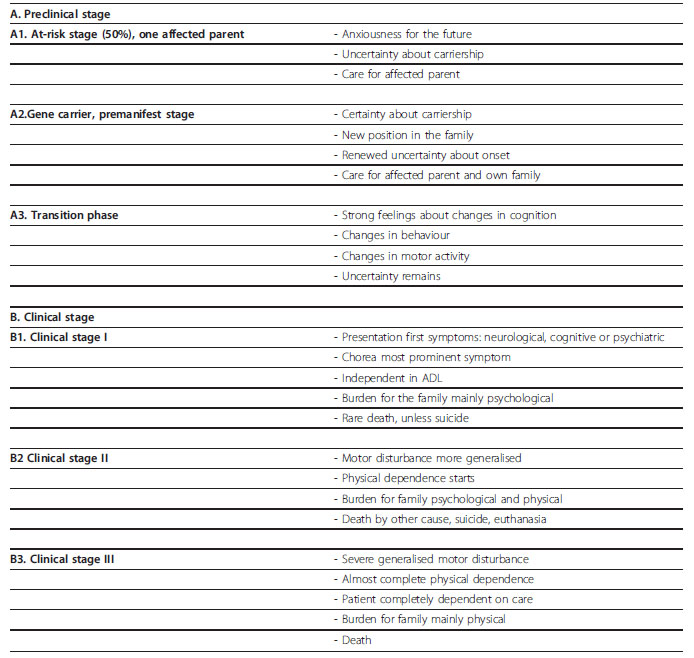
Tabla 1. Estadios del Síndrome de Hungtinton
Fuente: Roos (2010)
El SH es una enfermedad hereditaria autosómica dominante, causada por una elongación del trinucleotido CAG, en el cromosoma 4p16.3, en el gen Huntingtine (Hunington’s disease collaborative research group, 1993). Las manifestaciones clínicas pueden ocurrir si el número de repeticiones excede las 40 en la codificación por poliglutamina, en la proteína que va de rango 6-26 (Roos, 2010). Este fenómeno, es principalmente visto en la línea reproductiva masculina (Trottier Y, Biancalana V, & Mandel JL, 1994).
Cuando el SH inicia antes de los 20 años, se le conoce como la enfermedad de Huntington juvenil, en la que estas repeticiones exceden los 55 (Trottier Y, Biancalana V, & Mandel JL, 1994).
En la patogénesis en general encontramos una atrofia cerebral particularmente en el estriado, con una extensa perdida neuronal (Imarisio S, y otros, 2008).
Diagnóstico
Para determinar el diagnóstico adecuado del SH, nos basamos en los síntomas y signos clínicos de una persona en la que se confirme la herencia autosómica dominante por parte de su progenitor. Por lo tanto, en primer lugar, es necesaria la historia clínica donde consten antecedentes patológicos familiares (Roos, 2010)
El actual Gold Standard, es la determinación del ADN, que demuestre una repetición del CAG de al menos 36 en el gen Huntingtine, en el cromosoma 4 (Roos, 2010).
Estudios imagenológicos, como la tomografía computarizada y la resonancia magnética, muestran una disminución gradual de los ganglios basales y una atrofia de las cortezas frontal y temporal (Bates G, Harper P, & Jones L, 2002).
Diagnóstico Prenatal
El diagnóstico prenatal es posible entre las 10 y 12 semanas de embarazo, mediante un muestreo de las vellosidades coriónicas; y entre las 15 y 17 semanas, mediante amniocentesis. Los padres que conocen su estado genético, son a quienes se sugiere realizar estos procedimientos. El procedimiento se efectúa con la intención de culminar el embarazo, en caso de un resultado positivo para el gen Huntingtine en el embrión (Decruyenaere M, y otros, 2007).
Diagnóstico Diferencial
Para poder comparar con otra patología debemos considerar los síntomas motores y relacionarlo con las patologías que se expresan en la Tabla 2 (Roos, 2010).
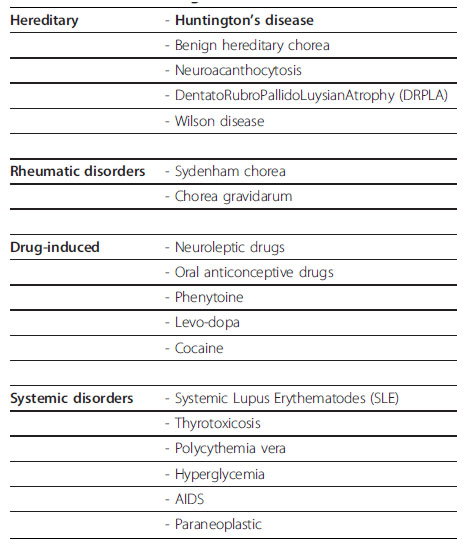
Tabla 2. Diagnóstico diferencial por corea
Fuente: Roos (2010)
Tratamiento
A pesar de que el SH es una enfermedad para la cual no existe hasta la actualidad una cura disponible, se han elaborado diversas opciones farmacológicas para el tratamiento de los síntomas y signos que este síndrome genera (Hamilton JM, y otros, 2003).
El tratamiento se emplea en la prescripción de drogas y medicamentos. La cirugía no tiene un rol importante en el SH (Roos, 2010).
Tratamiento farmacológico según los signos y síntomas clínicos
El signo clínico más relevante es la hiperquiinesia o corea, el cual es tratado con los bloqueantes de receptores de dopamina. Los fármacos más usados se dividen en neurolépticos típicos y atípicos, entre los que constan: Tiaprida, Olanzapina, Pimozida y Risperidona (Bonelli RM & Wenning GK, 2006). Por otro lado, como esta enfermedad tiene una afectación social de gran impacto, ocasiona depresión y comportamientos agresivos, para los que se emplean fármacos antidepresivos como: Citalopram, Fluoxetina y Mirtazapina; y para controlar el comportamiento agresivo se prescribe Sertralina y Olanzapina (Huntington Study Group, 2006).
Estos tratamientos se le otorgan a los pacientes para mejorar su calidad de vida, mas no representan una cura para la enfermedad.
Conclusión
El síndrome de Huntington (SH) es una enfermedad neurodegenerativa que, aunque no afecta en un porcentaje significativo a la población, aumenta su morbilidad y mortalidad, convirtiéndose en un problema para la salud pública. Tener conocimiento sobre sus manifestaciones clínicas y poder así conseguir un diagnóstico para ejecutar un buen tratamiento es muy importante, debido a que, al no existir una cura definitiva para esta enfermedad, brindar una mejor calidad de vida los pacientes que la padecen, se convierte en el objetivo principal de los médicos de atención primaria con el aporte de este artículo.
REFERENCIAS BIBLIOGRÁFICAS
Bates G, Harper P, & Jones L. (2002). Huntington’s disease. Oxford: Oxford University Press.
Bonelli RM, & Wenning GK. (2006). Pharmacological management of Huntington’s disease: an evidence-based review. Curr Pharm Des, 12, 2701-20.
Bruyn GW. (1968). Huntington’s chorea: historical, clinical and laboratory synopsis (Vol. 6). Amsterdam: Vinken PJ.
Conneally, P. M. (1984). Huntington disease: genetics and epidemiology. American Journal of Human Genetics, 506–26.
Decruyenaere M, Evers-Kiebooms G, Boogaerts A, Philippe K, Demyttenaere K, Dom R, . . . Vandenberghe W. (2007). The complexity of reproductive decision-making in asymptomatic carriers of the Huntington mutation. Eur J Hum Genet, 15, 453-62.
Hamilton JM, Salmon DP, Corey-Bloom J, & et al. (2003). Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry, 74, 120 –122.
Hunington’s disease collaborative research group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell, 72, 971-983.
Huntington Study Group. (2006). Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology, 66, 366-72.
Imarisio S, Carmichael J, Korochuk V, Chien-Wen Ch, Saiki S, Rose C, . . . Ttofi E. (2008). Huntington’s disease: from pathology and genetics to potential therapies. Biochem J, 412, 191-209.
Roos, R. A. (2010). Huntintong's disease a clinical review. Orphanet j Rare Dis, 5, 1-8. doi:https://doi.org/10.1186/1750-1172-5-40
Trottier Y, Biancalana V, & Mandel JL. (1994). Instability of CAG repeats in Huntington’s disease: relation to parental transmission and age of onset. J Med Genet, 31, 377-82.