
DOI: 10.26820/recimundo/4.(4).octubre.2020.399-410
URL: http://recimundo.com/index.php/es/article/view/917
EDITORIAL: Saberes del Conocimiento
Revista: RECIMUNDO
ISSN: 2588-073X
Tipo de Investigación: Artículo de Revisión
Código UNESCO: Ciencias Médicas
Paginas: 399-410
Verónica Lucía Hernández Nieto1; Cristian Daniel Chacón Molina2; Jonnathan Adrián Verdugo Illescas3; Jessica Lorena Chaves Diaz4
 https://orcid.org/0000-0003-2303-7570 https://orcid.org/0000-0003-1127-7227 https://orcid.org/0000-0002-7599-9061 https://orcid.org/0000-0003-1973-7988
https://orcid.org/0000-0003-2303-7570 https://orcid.org/0000-0003-1127-7227 https://orcid.org/0000-0002-7599-9061 https://orcid.org/0000-0003-1973-7988CORRESPONDENCIA
Verónica Lucía Hernández Nieto
vlhernandezn@gmail.com
Quito, Ecuador
La granulomatosis con poliangítis (GPA) o también conocida como Granolomatosis de Wegener, es un tipo de vasculitis sistémica que afecta los vasos pequeños, más comúnmente afectando el tracto respiratorio y los riñones. El diagnóstico se basa en criterios clínicos, pruebas de laboratorio, imágenes e histología. El noventa por ciento son ANCA (anticuerpos anticitoplasma de neutrofílico) positivos. La histología demuestra inflamación granulomatosa, necrosis y vasculitis, mientras que, los estudios de imágenes son vitales para la evaluación inicial y el seguimiento. De tal manera, la tomografía computarizada (TC) es el método de elección para la evaluación de la vía aérea superior y los pulmones, debido a su alta sensibilidad para detectar anomalías de los senos paranasales, el árbol bronquial y los pulmones. Aunado a ello, la resonancia magnética está indicada para la evaluación del sistema nervioso central y el corazón, sin embargo, la tomografía por emisión de positrones PET/TC tiene alta sensibilidad para enfermedades torácicas y abdominales, es útil para detectar lesiones que no se ven con otras técnicas de imagen y para el seguimiento. La afectación renal, muy frecuente en la GPA, suele ser indetectable en las imágenes, pero puede verse en la PET/TC en casos de glomerulonefritis marcada. No obstante, las radiografías simples no deben usarse para evaluar el GPA debido a su bajo rendimiento diagnóstico. Los pacientes son tratados con corticosteroides e inmunosupresores y deben llevar un seguimiento a largo plazo, ya que, generalmente las recaídas son frecuentes.
Palabras claves: Tomografía computarizada, Granulomatosis con poliangeítis, Tomografía por emisión de positrones, Granulomatosis de Wegener, vasculitis.
Granulomatosis with polyangiitis (GPA) or also known as Wegener's Granolomatosis, is a type of systemic vasculitis that affects small vessels, most commonly affecting the respiratory tract and kidneys. Diagnosis is based on clinical criteria, laboratory tests, images, and histology. Ninety percent are ANCA (anti-neutrophil cytoplasmic antibodies) positive. Histology demonstrates granulomatous inflammation, necrosis, and vasculitis, while imaging studies are vital for initial evaluation and follow-up. Thus, computed tomography (CT) is the method of choice for evaluating the upper airway and lungs, due to its high sensitivity to detect abnormalities of the paranasal sinuses, the bronchial tree, and the lungs. In addition to this, magnetic resonance imaging is indicated for the evaluation of the central nervous system and the heart, however, PET / CT positron emission tomography has high sensitivity for thoracic and abdominal diseases, it is useful to detect lesions that are not seen with other imaging techniques and for monitoring. Renal involvement, very common in GPA, is usually undetectable on imaging, but can be seen on PET / CT in cases of marked glomerulonephritis. However, plain radiographs should not be used to assess GPA due to their poor diagnostic performance. The patients are treated with corticosteroids and immunosuppressants and should be followed long-term, since relapses are generally frequent.
Keywords: Computed tomography, Granulomatosis with polyangiitis, Positron emission tomography, Wegener's granulomatosis, vasculitis.
Granulomatose com poliangiíte (GPA) ou também conhecida como Granolomatose de Wegener, é um tipo de vasculite sistêmica que afeta pequenos vasos, afetando mais comumente o trato respiratório e os rins. O diagnóstico é baseado em critérios clínicos, exames laboratoriais, imagens e histologia. Noventa por cento são ANCA (anticorpos citoplasmáticos anti-neutrófilos) positivos. A histologia demonstra inflamação granulomatosa, necrose e vasculite, enquanto os estudos de imagem são vitais para avaliação inicial e acompanhamento. Assim, a tomografia computadorizada (TC) é o método de escolha para avaliação das vias aéreas superiores e dos pulmões, devido à sua alta sensibilidade para detectar anormalidades dos seios paranasais, árvore brônquica e pulmões. Além disso, a ressonância magnética é indicada para avaliação do sistema nervoso central e do coração, porém a tomografia por emissão de pósitrons PET / CT tem alta sensibilidade para doenças torácicas e abdominais, é útil para detectar lesões que não são observadas com outras técnicas de imagem e para monitoramento. O envolvimento renal, muito comum no GPA, geralmente é indetectável na imagem, mas pode ser observado na PET / TC em casos de glomerulonefrite acentuada. No entanto, radiografias simples não devem ser usadas para avaliar GPA devido ao seu baixo desempenho diagnóstico. Os pacientes são tratados com corticosteroides e imunossupressores e devem ser acompanhados por longo prazo, pois as recidivas geralmente são frequentes.
Palavras-chave: Tomografia computadorizada, Granulomatose com poliangiite, Tomografia por emissão de pósitrons, Granulomatose de Wegener, vasculite.
INTRODUCCIÓN
Las vasculitis es un grupo de enfermedades caracterizadas por inflamación de las paredes de los vasos sanguíneos. David & Felipe, (2019) indican:
La granulomatosis de Wegener (WG) es una enfermedad autoinmune multisistémica rara de etiología desconocida, caracterizada por vasculitis granulomatosa inflamatoria y pauci-inmune necrotizante en vasos sanguíneos de pequeño y mediano tamaño (capilares, vénulas, arteriolas y arterias) asociada a anticuerpos anticitoplasma de neutrófilos (ANCAS) dirigida contra proteinasa 3 (PR3)”, una serina proteasa de neutrófilos, presentada en gránulos azurófilos primarios de neutrófilos polimorfonucleares (PMN) y lisosomas de monocitos (p.30).
Por lo general, produce inflamación granulomatosa de las vías respiratorias superiores e inferiores y una glomerulonefritis necrotizante segmentaria (tríada clásica de la enfermedad), aunque cualquier órgano puede estar afectado.
Este tipo de patología puede presentarse a cualquier edad con mayor incidencia entre los 65 y 74 años de edad. “Su incidencia anual es de 5-10/millón con una prevalencia de 24-157 casos por millón, siendo similar entre ambos sexos” (Shi, 2017). “Es extremadamente raro en los negros en comparación con los blancos; la proporción hombre: mujer es de 1: 1 y, aunque la enfermedad se puede observar a cualquier edad, la edad media de diagnóstico es de 40 años” (Carol & Langford Anthony, 2008).
Las características histopatológicas de la GW son vasculitis necrosante de vasos sanguíneos de pequeño y mediano tamaño con formación de granulomas y una importante tríada de inflamación granulomatosa, vasculitis y necrosis. “La biopsia pulmonar muestra la típica vasculitis granulomatosa necrotizante, casi siempre con nódulos múltiples y bilaterales (el 50% son cavitados) o infiltrados difusos, compuestos por neutrófilos, linfocitos, células plasmáticas, histiocitos y eosinófilos” (Comarmond & Cacoub, 2014).
El tejido del tracto respiratorio superior revela inflamación aguda y crónica, necrosis y lesiones granulomatosas, con o sin vasculitis, siendo la tríada de características patológicas presentes solo en alrededor del 15% de los casos.
La afectación renal se manifiesta por glomerulonefritis necrotizante focal y segmentaria, a menudo con semilunas e infiltrados tubulointersticiales mononucleares, sin evidencia de depósito de inmunocomplejos (pauci-inmunes en inmunofluorescencia o microscopía electrónica). Sin embargo, los granulomas se encuentran con poca frecuencia en las muestras de biopsia renal (Haas & Eustace, 2004).
La inmunopatogénesis de esta enfermedad aún no está clara, aunque el conocimiento ha experimentado un progreso sustancial en los últimos años, y se cree que está involucrada la inmunidad tanto celular como humoral. Además, los antecedentes genéticos y los factores ambientales pueden desempeñar un papel importante en WG. De tal manera durante el desarrollo del proceso investigativo, se presenta una revisión bibliográfica con el objetivo de brindar información de interés que sirva de guía para futuras investigaciones.
Metodología
Para el desarrollo de este proceso investigativo, se plantea como metodología la encaminada hacia una orientación científica particular que se encuentra determinada por la necesidad de indagar en forma precisa y coherente una situación, en tal sentido Davila, (2015) define la metodología “como aquellos pasos anteriores que son seleccionados por el investigador para lograr resultados favorables que le ayuden a plantear nuevas ideas” (p.66)
Lo citado por el autor, lleva a entender que el desarrollo de la acción investigativa busca simplemente coordinar acciones enmarcadas en una revisión bibliográfica con el fin de complementar ideas previas relacionadas Enfermedad de Wegener. Tratamiento y diagnóstico a través de una revisión de literatura, para así finalmente elaborar un cuerpo de consideraciones generales que ayuden a ampliar el interés propuesto.
Tipo de Investigación
Dentro de toda práctica investigativa, se precisan acciones de carácter metodológico mediante las cuales se logra conocer y proyectar los eventos posibles que la determinan. En este sentido, la presente investigación corresponde al tipo documental, definido por Castro (2016), “se ocupa del estudio de problemas planteados a nivel teórico, la información requerida para abordarlos se encuentra básicamente en materiales impresos, audiovisuales y / o electrónicos”. (p.41).
En consideración a esta definición, la orientación metodológica incluye la oportunidad de cumplir con una serie de actividades inherentes a la revisión y lectura de diversos documentos, donde se encuentran ideas explicitas relacionadas con los tópicos encargados de identificar una característica inmersa en el estudio. Por lo tanto, se realizaron continuas interpretaciones con el claro propósito de revisar aquellas apreciaciones propuestas por diferentes investigadores en relación al tema de interés, para luego dar la respectiva argumentación a los planteamientos, en función a las necesidades encontradas en la investigación, apoyados en las herramientas tecnológicas para la búsqueda de trabajos con valor científico disponibles en la web que tenían conexión con el objetivo principal de la investigación.
Fuentes Documentales
El análisis correspondiente a las características que predomina en el tema seleccionado, llevan a incluir diferentes fuentes documentales encargadas de darle el respectivo valor científico y en ese sentido cumplir con la valoración de los hechos a fin de generar nuevos criterios que sirven de referencia a otros procesos investigativos. Para Castro,(2016) las fuentes documentales incorporadas en la investigación documental o bibliográfica, “representa la suma de materiales sistemáticos que son revisados en forma rigurosa y profunda para llegar a un análisis del fenómeno” (p.41). Por lo tanto, se procedió a cumplir con la lectura previa determinada para encontrar aquellos aspectos estrechamente vinculados con el tema, con el fin de explicar mediante un desarrollo las respectivas apreciaciones generales de importancia.
Técnicas para la Recolección de la Información
La conducción de la investigación para ser realizada en función a las particularidades que determinan a los estudios documentales, tiene como fin el desarrollo de un conjunto de acciones encargadas de llevar a la selección de técnicas estrechamente vinculadas con las características del estudio. Bolívar, (2015), refiere, que es “una técnica particular para aportar ayuda a los procedimientos de selección de las ideas primarias y secundarias”. (p.71).
Tal como lo expresa, Bolívar, (2015) “Las técnicas documentales proporcionan las herramientas esenciales y determinantes para responder a los objetivos formulados y llegar a resultados efectivos” (p. 58). Es decir, para responder con eficiencia a las necesidades investigativas, se introdujeron como técnica de recolección el método inductivo, que hizo posible llevar a cabo una valoración de los hechos de forma particular para llegar a la explicación desde una visión general. El autor Bolívar, (2015) tambien expresa que las técnicas de procesamiento de datos en los estudios documentales “son las encargadas de ofrecer al investigador la visión o pasos que deben cumplir durante su ejercicio, cada una de ellas debe estar en correspondencia con el nivel a emplear” (p. 123). Esto indica, que para llevar a cabo el procesamiento de los datos obtenidos una vez aplicadas las técnicas seleccionadas, tales como: fichas de resumen, textual, registros descriptivos entre otros, los mismos se deben ajustar al nivel que ha sido seleccionado.
Resultados
Es común ver que la WG progresa de una etapa localizada, restringida al tracto respiratorio, a una enfermedad sistémica (generalizada), y esta transición puede estar influenciada por una serie de factores de riesgo genéticos. “Existe evidencia de que el sistema de antígeno leucocitario humano (HLA) está involucrado tanto en el desarrollo de WG como en otras vasculitis positivas para ANCA” (Jagiello, Gencik, & Arning, 2004). La medicina trabaja fuertemente en investigaciones para confirmar esta asociación, por ejemplo, para confirmar la posible relación entre WG y una región en el cromosoma 6p21.3, aunque se necesita de más estudios.
Esta enfermedad autoinmune está genéticamente asociada con la región de clase II del complejo principal de histocompatibilidad (MHC), como el alelo HLA-DPB1 * 0401
De manera similar a otras enfermedades autoinmunes, el polimorfismo funcional, 620W, en el gen de la tirosina fosfatasa intracelular PTPN 22, es un factor predisponente para WG en presencia de ANCA. Esto se debe a la pérdida de función de la proteína PTPN 22, que es importante en la señalización del receptor de células T (TCR), a través de la inhibición de moléculas clave en la vía del receptor, lo que resulta en la activación y proliferación de las células T y, posteriormente, en las reacciones humorales. alteraciones. (Jagiello, Gencik, & Arning, 2004)
Otro factor de riesgo genético para la WG es el polimorfismo CTLA-4 (antígeno 4 de linfocitos T citotóxicos).La exposición ambiental a solventes o sílice, la agricultura y la vida en latitudes del norte se ha asociado con el desarrollo de esta enfermedad autoinmune 0401.
Existe una relación entre el WG y las enfermedades infecciosas. “El transporte nasal crónico de Staphylococcusaureus (la frecuencia en una población sana varía del 20 al 35%) se ha asociado con una tasa de recaída más alta de WG, lo que sugiere un papel de estas bacterias en su fisiopatología” (David & Felipe, 2019). Varias toxinas de este patógeno estimulan la actividad de las células B y probablemente de las células T. Además, se ha documentado la estimulación directa de neutrófilos in vitro, con Staphylococcusaureus produciendo proteinasas con actividad contra inhibidores de proteinasas humanas, como la α1-antiproteasa, que es el principal inhibidor de proteinasa-3, lo que resulta en una actividad inflamatoria persistente.
Manifestaciones clínicas
La enfermedad de Wegener tiene un espectro de presentaciones clínicas. Mientras la enfermedad está activa, la mayoría de los pacientes presentan síntomas y signos inespecíficos como malestar, debilidad, artralgias migratorias, anorexia, pérdida de peso, sudores nocturnos y fiebre. Es importante excluir una infección secundaria cuando hay fiebre. Estos síntomas constitucionales pueden durar de semanas a meses sin evidencia de afectación de órganos específicos.
Oído, nariz y garganta Las manifestaciones (ORL) son frecuentes en WG. “La afectación de las vías aéreas superiores ocurre en el 95% de los pacientes, siendo la sinusitis crónica la queja inicial más común (67%)” (Jayne, 2009). Otras manifestaciones frecuentes incluyen deformidad de la nariz en silla de montar por colapso del soporte nasal con destrucción de hueso y cartílago, estenosis traqueal subglótica (16%) que causa estridor, rinitis, epistaxis, úlceras orales y / o nasales, otitis media serosa, otorrea y pérdida auditiva (conductiva y neurosensorial). (Fig. Nº1)
Compromiso pulmonar también es común y puede manifestarse como infiltrados pulmonares “difusos asintomáticos (71%), nódulos, atelectasias, consolidación y / o derrame pleural o puede expresarse clínicamente como tos (34%), hemoptisis (18%) debido a bronquiectasias, cavitación pulmonar lesiones parenquimatosas o hemorragia alveolar difusa (HAD), disnea (7%), dolor pleurítico y malestar torácico (8%)” (Manganelli, Fietta, & Carotti, 2006). También puede ocurrir el desarrollo de fibrosis pulmonar e hipertensión arterial pulmonar. La radiografía de tórax y la tomografía computarizada (TC) son investigaciones importantes que deben incluirse en el diagnóstico de estos pacientes.
La GW también se puede reconocer como masas tumorales fuera del pulmón, siendo la mama y el riñón las localizaciones extratorácicas más comunes, aunque estos casos son raros. Otra complicación típica es la enfermedad renal, que se presenta en un 17% al diagnóstico inicial y en un 77% a lo largo de la evolución de la enfermedad, manifestándose con insuficiencia renal aguda, hematuria, cilindros eritrocitarios y proteinuria.
Participación ocular “(52%) se manifiesta como conjuntivitis, dacriocistitis, queratitis, uveítis, escleritis, vasculitis del nervio óptico, oclusión de la arteria retiniana, diplopía y proptosis por masa retro orbitaria” (Harper, Letko, & Samson, 2001).
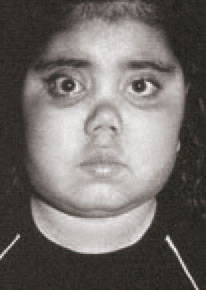
Figura 1. Paciente con granulomatosis de Wegener con proptosis de ojo derecho y deformación del tabique nasal (nariz en silla de montar).
Fuente: (Olga Vera & Arturo Olvera, 2009).
Manifestaciones cutáneas, presentes en el 45% de los pacientes, son hallazgos inespecíficos y suelen afectar a las extremidades inferiores. “Púrpura palpable, pápulas, nódulos subcutáneos, ulceraciones, livedoreticularis y urticaria son frecuentes, siendo la angitisleucocito clástica (púrpura, necrosis focal y ulceración) la lesión cutánea más frecuente” (Merkel, Lo, & Holbrook, 2005).

Figura 1. Paciente con granulomatosis de Wegener donde se muestran las piernas con lesiones dérmicas caracterizadas por úlceras y costras.
Fuente: (Olga Vera & Arturo Olvera, 2009).
Sistema musculoesqueléticoTambién suele estar implicado por el proceso vasculítico, con “mialgias, artralgias poliarticulares y simétricas, y artritis no deformantes de las grandes articulaciones, estando presente en el 32% de los pacientes al diagnóstico inicial y en el 67% a lo largo de la evolución de la enfermedad” (Merkel, Lo, & Holbrook, 2005).
Manifestaciones cerebrales “(23%) incluyen neuropatía de pares craneales, vasculitis cerebral, mono neuritis múltiple, Paqui meningitis, lesiones masivas del sistema nervioso central, entre otras manifestaciones menos frecuentes. Sin embargo, la neuropatía periférica es más común que la afectación del sistema nervioso central” (Graham, 2014).
En pacientes con WG se ha documentado una alta incidencia de eventos trombóticos venosos (TEV). Según el estudio Wegener's Clinical Occurrence of Thrombosis (WeCLOT).
La incidencia de TEV fue de 7/100 personas-año, pero no se recomienda la anticoagulación rotina para todos los pacientes. Los anticuerpos anti plasminógeno fueron más altos en los pacientes con PR3-ANCA que en los controles sanos (cinco de cada nueve pacientes con TEV fueron positivos para anticuerpos anti plasminógeno). (Merkel, Lo, & Holbrook, 2005, p.620)
Con menor frecuencia, afectación del corazón (presente en el 6-44% de los pacientes con pericarditis, miocarditis, anomalías del sistema de conducción y vasculitis coronaria); tracto gastrointestinal (vasculitis esplácnica, dolor abdominal); cavidad oral ("hiperplasia gingival de fresa"); Se observa tracto genitourinario inferior (uretra, uréteres, cérvix, vagina, testículo y próstata), glándulas parótidas, glándula tiroides (hipertiroidismo), hígado o mama.
A continuación se presenta en una tabla donde se observa el porcentaje de afectación de los órganos de acuerdo con el avance de la enfermedad.
Tabla 1. Porcentaje de afectación de órganos en WG
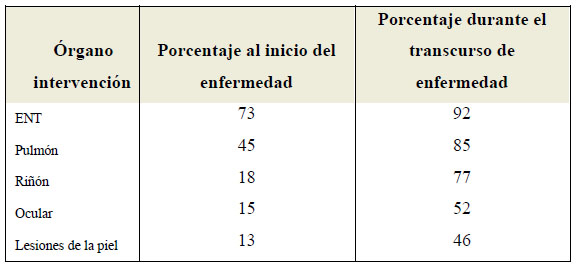
Fuente: (Hoffman, Kerr, & Leavitt, 1992)
n la tabla se puede observar que los órganos de la nariz el oído y la garganta (ENT) de acuerdo a sus siglas en ingles, son los más afectados a medida que la enfermedad avanza con un porcentaje del 92%, seguido del pulmón, riñón y las lesiones oculares, siendo la menos marcada las lesiones en la piel.
Diagnóstico
En un paciente con las características clínicas descritas anteriormente, el diagnóstico de WG requiere la demostración de vasculitis granulomatosa necrotizante en la biopsia de tejido, a menudo de un sitio de enfermedad activa, siendo las biopsias de pulmón y riñón las más específicas (la biopsia de pulmón se realiza en ausencia de afectación renal).
El WG se puede clasificar según la nomenclatura de la Conferencia del Consenso de Chapel Hill (CHC) para vasculitis sistémica primaria (tabla 2) y según los criterios de clasificación definidos por el American College of Rheumatology (ACR) (tabla 3) en 1990. (Jennette, Falk, & Andrassy, 1994)
Tabla 2. Definición de WG según la Conferencia de Consenso de Chapel Hill

Fuente: (Jennette, Falk, & Andrassy, 1994)
Tabla 3. Criterios de clasificación del American College of Rheumatology

Fuente: (Leavitt, Fauci, & Bloch, 1990)
El European Vasculitis Study Group (EUVAS) ha desarrollado una definición del estadio de la enfermedad que incluye la enfermedad localizada (Jayne D. , 2001). El WT también se puede dividir en enfermedad limitada (ausencia de características de la enfermedad que representen una amenaza inmediata para un órgano individual crítico o para la vida del paciente) o enfermedad grave.
Diagnóstico diferencial
El diagnóstico diferencial incluye patologías con características clínicas relacionadas, principalmente síndrome pulmón-riñón, o con hallazgos de laboratorio similares (ANCA positivos). La siguiente tabla enumera los trastornos más relevantes.
Tabla 4. Diagnóstico diferencial de WG
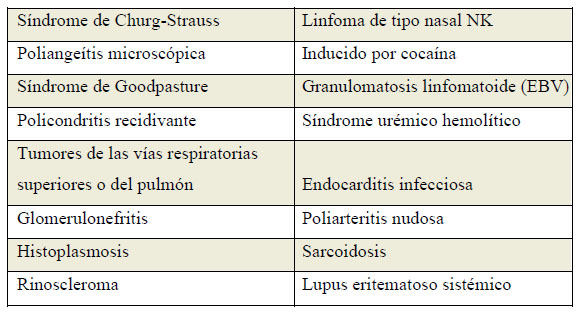
Fuente: (Leavitt, Fauci, & Bloch, 1990)
Tratamiento
La terapia se adapta según el estadio y la actividad de la enfermedad y requiere inducción de la remisión con terapia inmunosupresora inicial, seguida de terapia inmunosupresora de mantenimiento para prevenir la recaída y controlar la enfermedad.
Inducción a la remisión: “Los pacientes con enfermedad limitada (localizada / sistémica temprana) deben ser tratados con esteroides orales 1 mg / kg / día en combinación con metotrexato oral o subcutáneo (20-25 mg / semana), según el estudio” (Stone, Hoffman, & Merkel, 2001). Se recomienda ácido fólico diario 1 mg / día. En estos casos, se debe evitar la ciclofosfamida debido a la toxicidad asociada. El metotrexato es igual a la ciclofosfamida en la inducción de remisión, aunque el primero se asoció con más recaídas a los 18 meses, en comparación con la ciclofosfamida, 69,5% vs 46,5%, respectivamente.
Los pacientes con enfermedad generalizada o enfermedad grave (función de órganos vitales amenazada) deben tratarse con ciclofosfamida en combinación con esteroides (esquema FAUCI, introducido en la década de 1970).
La ciclofosfamida se puede administrar por vía oral (2 mg / kg / día y la dosis máxima es de 200 mg / día) o por vía intravenosa (15 mg / kg cada 2 semanas durante los primeros 3 pulsos, luego cada 3 semanas durante los siguientes 3-6 pulsos, siendo el máximo dosis 1500 mg) y debe ajustarse a la función renal y la edad (tabla 6). La duración total del tratamiento no debe exceder los 6 meses. La ciclofosfamida pulsada tiene menos efectos secundarios (menor tasa de leucopenia) que las dosis orales, con la misma eficacia para la inducción de la remisión. (Stone, Hoffman, & Merkel, 2001).
Tabla 5. Reducciones pulsadas de ciclofosfamida para la función renal y la edad

Fuente: (Stone, Hoffman, & Merkel, 2001)
Los esteroides siempre se consideraron la piedra angular de la terapia. Aunque su uso no se ha evaluado en ensayos controlados aleatorios, todos los ensayos clínicos han utilizado esta terapia en combinación con otro inmunosupresor. Se desconoce la dosis y la duración ideales de la terapia con esteroides y se necesitan más estudios. Suelen administrarse por vía oral (1 mg / kg / día de prednisolona durante 1 mes y luego disminuyendo lentamente), pero en el conjunto de glomerulonefritis rápidamente progresiva y / o hemorragia alveolar pulso intravenoso metilprednisolona (0,5-1 g / día durante 3 días consecutivos) puede ser usado. En estos dos entornos, la plasmaféresis también se puede considerar como adyuvante, aunque no se ha demostrado que mejore la supervivencia global. “Los pacientes tratados con ciclofosfamida y corticoides deben recibir profilaxis frente a la neumonía por Pneumocystisjiroveci con trimetoprim / sulfametoxazol 960 mg 3 veces por semana o, en caso de alergia a este antibiótico, 100 mg diarios de dapsona” (Stone, Hoffman, & Merkel, 2001).
Mantenimiento de remisión: Para minimizar la exposición a los efectos secundarios del tratamiento con ciclofosfamida, en los últimos años se han propuesto terapias alternativas. En el mantenimiento de la remisión se pueden utilizar tres agentes: azatioprina (2 mg / kg / día), metotrexato (20-25 mg / semana) y leflunomida. Según el ensayo CYCAZAREM Pagnoux, Mahr, & Hamidou, (2008), el primero es más seguro y tan eficaz como la ciclofosfamida.
El micofenolato de mofetilo (2 g / día) también se puede considerar como terapia de mantenimiento cuando existe intolerancia o falta de eficacia con azatioprina o metotrexato, aunque se necesitan más estudios.
Terapias alternativas. Las terapias alternativas se han investigado cada vez más, algunas de ellas con resultados prometedores. Varios estudios mostraron mejoría clínica o remisión con rituximab (375 mg / m2 por semana durante cuatro semanas).
Estos datos fueron confirmados por dos ensayos recientes, RAVE y RITUXVAS, que mostraron la no inferioridad de rituximab en comparación con la ciclofosfamida para la inducción de la remisión para el primer estudio y la remisión de la enfermedad grave (afectación renal) para el segundo ensayo, con el estudio anterior que informó una posible superioridad de rituximab en la enfermedad recidivante y en el ensayo RITUXVAS el grupo de rituximab se asoció con una alta tasa de eventos adversos graves (Wegener´s Granulomatosis Etanercept Trial (WGET) Research Group, 2005).
Los estudios de etanercept, a saber, el ensayo de granulomatosis etanercept de Wegener (WGET) no encontraron mejoría en el mantenimiento de la remisión, con actividad de la enfermedad grave o no potencialmente mortal, cuando se agregó a la terapia estándar, con un posible riesgo de aumentar las infecciones y malignidades. Este estudio no evaluó este fármaco en sí.
El infliximab, las globulinas antitimocíticas y el alentuzumab (estudio CAMPATH-1H) están bajo investigación y se necesitan más estudios para definir su función en la terapia. Algunas investigaciones sugieren el uso de inmunoglobulina intravenosa para la vasculitis refractaria, cuando la terapia convencional está contraindicada o para la enfermedad recidivante. Sin embargo, se necesitan más estudios.
Conclusión
Durante el desarrollo del proceso investigativo se observó que la enfermedad de Wegener es una enfermedad crónica grave que generalmente produce recaída en los pacientes que lo padecen. No obstante, su pronóstico ha mejorado con la aplicación de terapia prolongada y adaptada. Uno de los principales problemas terapéuticos es la aparición de efectos secundarios relacionados con el tratamiento, cuya gravedad y frecuencia se asocia con una prolongación del tratamiento, que sigue siendo difícil de evitar.
Por tal manera se considera que los pacientes con WG deben tener un seguimiento de cerca, con visitas regulares a su médico, que involucre un enfoque multidisciplinario y exámenes frecuentes para monitorear la enfermedad. Esto debido a que las recaídas se consideran comunes en un cincuenta por ciento de los pacientes justo a los cinco años de haber padecido la enfermedad.
Esta patología, generalizada o grave no tratada, tiene un pronóstico más precario por lo que es importante que una vez diagnosticada se proceda a administrar tratamiento y terapia inmunosupresora para aumentar la vida útil de los pacientes debido a que la tasa de mortalidad es bastante marcada cuando no se trata luego de dos años. Sin embargo, cuando el paciente presenta complicación renal es probable que ya se encuentre en estado crítico. Para este tipo de patologías las principales causas de muertes se deben a infección, insuficiencia respiratoria y renal, malignidad y enfermedad cardiovascular, siendo la primera la principal responsable de la mortalidad y morbilidad en WG. Finalmente se puede concluir que, es de vital importancia realizar el tratamiento indicado tomando en consideración que el mantenimiento de remisión y el seguimiento por parte del paciente y del médico tratante debe ser lo más relevante para evitar las recaídas frecuentes que se generan con el paso de los años.
REFERENCIAS BIBLIOGRÁFICAS
Carol, A., & Langford Anthony, S. (2008). Fauci. The vasculitis syndromes. In: Fauci A. et all. Harrison´s – Principles of Internal Medicine; 17th edition. Mc Graw Hill, 2119-2124.
Comarmond, C., & Cacoub, P. (2014, Agosto 20). Granulomatosis with polyangiitis (Wegener): Clinical aspects and Conference . Autoimmun Rev, 13, 1121-1125.
David, G., & Felipe, C. (2019). Actualización en el estudio de Granulomatosis con poliangeitis (Granulomatosis de Wegener). Rev Chil Radiol , 25(1), 26-34.
Graham, K. (2014). Vasculitic neuropathies. (Vol. 13). Lancet Neurol.
Haas, M., & Eustace, J. (2004). Immune complex deposits in ANCA-associated crescentic glomerulonephritis: a study of 126 cases. Kidney Int , 2145.
Harper, S., Letko, E., & Samson, C. (2001). Wegener´s granulomatosis: the relationship between ocular and systemic disease. J Rheumatol , 1025.
Hoffman, G., Kerr, G., & Leavitt, R. (1992). Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med, 116:488.
Jagiello, P., Gencik, M., & Arning, L. (2004). New genomic region for Wegener´s granulomatosis as revealed by an extended association screen with 202 apoptosis related genes. Hum Genet, 468-77.
Jayne, D. (2001). Update on the European Vasculitis Study Group trials. Current Opinion in Rheumatology, 13(1), 48-55.
Jayne, D. (2009). The diagnosis of vasculitis. Best Practice & Research Clinical Immnunology , The diagnosis of vasculitis.
Jennette, J., Falk, R., & Andrassy, K. (1994). Nomenclature of systemic vasculitides. Proposal of an internacional consensus conference. Arthritis Rheum, 187-92.
Leavitt, R., Fauci, A., & Bloch, D. (1990). The American College of Rheumatology 1990 criteria for the classification of Wegener´s granulomatosis. Arthritis Rheum , 1101-7.
Manganelli, P., Fietta, P., & Carotti, M. (2006). Respiratory system involvement in systemic vasculitis. Clin Exp Rheumatol. March-April, S48-S59.
Merkel, P., Lo, G., & Holbrook, J. (2005). Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener´s Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med , 620.
Moosig, F., Lamprecht, P., & Gross, W. (2008). Wegener´s Granulomatosis: the current view. Clin Rev Allergy Immunol, 19-21.
Olga Vera, L., & Arturo Olvera, A. (2009). Granulomatosis de Wegener, abordaje diagnóstico y terapéutico. Gac Méd Méx, 146(2), 1-9.
Pagnoux, C., Mahr, A., & Hamidou, M. (2008). Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med, 2790-2803.
Phillip, R., & Lugman, R. (2008). Mortality in systemic vasculitis: a systematic review. Clin Exp Rheumatol, S94-S104.
Shi, L. (2017). Anti-neutrophil cytoplasmic antibody-associated vasculitis: prevalence, treatment, and outcomes. Rheumatol Int. , 37(11), 1779-1788.
Stone, J., Hoffman, G., & Merkel, P. (2001). A disease-specific activity index for Wegener´s Granulomatosis: modification of the BVAS. Arthritis & Rheumatism , 912-920.
Wegener´s Granulomatosis Etanercept Trial (WGET) Research Group. (2005). N Engl J Med, 351-361.